| 疾病中文名 | 肾上腺脑白质退化症 | 疾病英文名 | Adrenoleukodystrophy |
| 别名 | 肾上腺脑白质失氧症/过氧化体症 | 患病率 | 1-9 / 100 000 |
| 遗传方式 | X连锁隐性 | ICD编码 | E71.3 |
疾病分类 先天性代谢异常 矿物离子缺陷
病因
Haberfeld and Spieler在1910年发表第一位患有ALD症状的病人是一位六岁的男孩,他视力减退、变的冷漠、学业退步;四个月后,他的步伐变的蹒跚、无法行走;七岁时住进医院,身上皮肤黝黑;八个月后身亡。他的另一个哥哥在八岁五个月大时,因为相同的症状死亡。Schilder 在1913年研究三个有相同状况的小孩脑部时,发现他们的大脑半球已经糜烂、没有神经髓鞘、同时布满了节结。Siemerling and Creutzfeldt (1923)发现患有此病时,他们的肾上腺同时也受到了破坏。于是在1970年,Blaw正式将此种疾病命名为肾上腺脑白质退化症(Adrenoleukodystrophy;ALD)。1997年,证实此种疾病是一种脂肪储存异常的疾病,患者无法有效的利用极长链脂肪酸(very long-chain fatty acids, VLCFA),造成脂肪在体内堆积而引起的疾病,因此又被称为过氧化体症(peroxisome disorder)(Moser,1997)。因患者体内细胞的过氧化体(peroxisome)在极长链脂肪酸的代谢中产生异常,尤其是C24、C26极长链脂肪酸异常堆积,极长链脂肪酸因此会堆积在大脑的白质和肾上腺的皮质内,进而侵蚀患者脑神经系统的髓鞘质,造成患者的髓鞘脱失,也算是一种发炎反应,脑部的神经细胞因此就会被摧毁,进而妨碍神经的传导。
1963年,Fanconi 观察Addison disease(肾上腺疾病)和Cerebral sclerosis(脑硬化)的个案时,发现几乎都是男性的病患或是母亲这一方的娘家舅舅也有相同症状的个案,因此而认定这是一种X染色体隐性遗传模式的神经退化疾病(Migeon,1981)。1976年,Igarashi 等人有一重大发现,他们在ALD 病人身上找到含有24~30个碳原子的高浓度极长链脂肪酸堆积在病人的大脑白质中的胆固醇脂和肾上腺内(Igarashi et al.,1976)。这意味着:这些堆积物很可能就是影响中枢神经系统内髓鞘的形成,甚至还有肾上腺皮质类酯醇生成的物质。
ALD 是一种饱和的极长链脂肪酸(含有24~ 30个C的长链分子,尤其是C26的分子 hexacosanoate)无法被分解断裂,于是堆积在大脑白质的胆固醇脂、肾上腺皮质内、或是在大脑鞘脂内。当然,在皮肤纤维母细胞内也可以找到极长链脂肪酸酸堆积的证据,利用这种方法可以拿来做为产前诊断或是探讨疾病致病机制的原理 (Moser et al., 1980)。既然这是一种极长链脂肪酸的代谢异常疾病,若是在饮食上加以限制,对于ALD 个案应该是有帮助的 (Moser, 1980)。
Moser(2000)依据ALD个案发病的年龄,还有他们在神经临床上的表现,将ALD分成了七种表现型:幼儿型脑白质化(childhood cerebral form)、肾上腺髓鞘神经型疾病(adrenomyeloneuropathy;AMN)、成人大脑型(adult cerebral)、青春期型(adolescent)、肾上腺病变型(Addison Only;adrenal insufficiency without neurological disease)、尚未发病之男性(asymptomatic)、女性异基因型(heterozygote),后来又发现女性异基因型的个案也同样会发病,因此将其定义为女性发病型 (female heterozygote) ,因为,在1991年,Dr. Moser研究900个男性病人及1000个带原女性发现有百分之五十的男性会在幼年或adolescent期病情会加速;百分之二十五的男性瘫痪的速度缓慢;少数的病人只有肾上腺的疾病(Addison disease),没有其他任何神经的症状;将近有百分之十五的女性带原者会产生渐进的强质性下半身瘫痪(Moser,2000)。
依临床的表现型来区分ALD个案的比例,大致上是:31~35%的儿童型脑白质化(childhood cerebral form)个案;40~46%是肾上腺脊髓性神经病变型(adrenomyeloneuropathy;AMN);14%为单纯的肾上腺病变型(adrenal insufficiency without neurological disease;Addison only);13%属于症状发生前之男性(asymptomatic);4~7%是青少年大脑型(Adolescent cerebral ALD);2~5%的成人大脑型(adult cerebral);和非常少的女性异合子病征基因型(Female Symptomatic Heterozygote)的个案(Moser,2000)。
患者依不同时间发病所表现的临床症状也很分歧,以最典型的儿童大脑型而言,平均发病年龄为7岁,会先丧失行动自主能力,而后语言能力渐也丧失,在一年、二年之内进入植物人状态,通常在诊断后数年内死亡。
1. 儿童型脑白质化(childhood cerebral form):
发生率约占所有类型的31~35%,发病年龄通常于4~8岁,很少会超过15岁。一旦开始发病通常表现出学习或行为异常,例如学校表现退步、阅读困难、方向感障碍、视觉及听力障碍,有时会出现复视及癫痫。通常发病后6个月到2年内会迅速地退化,即使发病年纪从2个月到20岁不等,逐渐丧失神经自主及运动能力。
2. 青少年大脑型 (Adolescent cerebral ALD):
一小部分的病人4~7%会在11到21岁发病,除了病情进展速度会比较慢一点外,其他症状大部分与儿童大脑型差不多。
3. 肾上腺脊髓性神经病变型(Adrenomyeloneuropathy, 简称AMN):
此型约占所有类型的40~46%,发生于在20岁至中年的成年人,主要症状为渐进式腿部僵硬与无力,无法控制括约肌,并伴随性功能的障碍,约10-20%的病患会因脑部的退化而有严重的认知及行为障碍,并会恶化到完全失能及丧失生命。
4. 女性异型合子病征基因型(Female Symptomatic Heterozygote):
ALD是性染色体病遗传疾病,代表着带因的女性通常带有一条好的x染色体,与一条有缺陷的x染色体。一般来说因为具有一条正常的x染色体,所以女性身上通常是不会见到病征。但是事实上还是有在女性身上发现ALD的病征,症状从轻微到严重都有,因此带因女性也需要注意。
在同一家族内表现出来的临床症状常常会有很大的差异性。譬如同一家族内的一位男性手足可能在幼儿期就已发病,可是他的另一位男性手足的临床症状就有可能是完全的正常;或是在一个家族中有四个病人,就有两种发病的型态:一位是在13岁时发病,另一位个案却是在41岁时才有症状(Davis et al.,1979);或是同一家族中一位是幼儿期发病,另一个案是青春期发病(Willems et al,1990);或是发现在同卵双生的男性中表现出不同类型的临床症状(Korenke et al.1996; Di Rocco et al. 2001) 。因此临床医师研究发展出一套方法检测和确认患者及带原者血液中VLCFA值(Moser et al.,1981);在收集了3,000个病人及29,000个正常对照组的检体结果发现:以VLCFA在新生儿阶段稍有上升为条件;对于男性患者的检验结果有非常高的准确度。有百分之八十五的带原者VLCFA值较高(Moser et al. 1999)。但是我们仍然无法从生化检验的异常结果或是从基因的突变位置上去预测这些个案的临床表征。
症状
ALD 病人常会被误诊为癫痫(epilepsy)、精神病(psychosis)、一种自闭症(亚斯伯格症,Asperer Syndrome)、或是一种脑肿瘤(brain tumor)(常会在手术后发现诊断错误)。有几项的临床症状可以当成鉴别诊断和参考依据,如:在早期常会因眼部神经的影响引起视力的退化;接着走路摇摆、手写字体变丑、然后说话不清楚;再接着的变化是会出现明显身体症状,如:抽筋、皮层性耳聋;最后是晚期的无法吞咽。成人型的ALD 也很容易被误诊为多发性硬化(Multiple Sclerosis)、强直性下半身轻瘫(Spastic Para paresis)、腰椎狭窄(Lumbar Stenosis)。所以,家族性的筛检诊断是非常重要的,可以尽早诊断出家族内的女性异基因型或是那些「尚未发病」的病人们,及早的做一些预防性的医疗准备。
曾有研究指出,被误诊后证实是幼儿型的ALD个案兄弟,老大十二岁被诊断为ADHD「注意力不集中、好动」,在ALD症状出现前,药物对增加注意力有进步,可是两年后个案开始变的话多、坐不住、冲动性;九岁的弟弟则表现在视力衰退、学习退步。最后才被证实罹患了ALD(Ievers et al.,1999)。这两位兄弟在做心理评估时表现出非常明显的症状,如:短期的记忆力、听力能力的障碍、要求重复问题、焦躁、坐不住、不听从指示。同时在同侪关系中适应不良、在阅读和数理方面的学习不良。父母对于两兄弟的评分同样是在注意力的不集中、坐不住、冲动、和不成熟的行为表现等(Ievers et al.,1999)。
对ALD个案的脑部电脑断层的解读,发展出一个疾病评分的标准,称之为Loes Score:针对个案脑部MRI看到的T2-weighted,因为在男性患者的身上可以看到一些行为异常时,个案脑部的MRI也可以看到T2的变化。有经验的神经学专家可以将Loes Score分成0~34分;MRI影像图的Loes Score分数愈高,表示脑内去髓鞘化白质化的程度愈严重(Loes et al.,2003)。
(病发初期) | (病程发展) |
活动过度 hyperactivity | 学习失能learning disabilities |
治疗
罗伦佐的油(Lorenzo's Oil) 中含有高量单元不饱和脂肪酸,是对于生化检验有异常,但神经症状尚未出现的ALD患者,最普遍被采用的治疗方式,以期减缓神经症状出现的速度(长期服用「罗伦佐的油」有百分之四十的机率,会出现血小板下降的问题)。其处方为:患者严格控制脂肪摄取,每公斤体重给予1.7公克glycerol trioleate oil(GTO)和0.3公克glycerol trierucate oil(GTE), 以取代一般油脂中饱和的极长链饱和脂肪酸。
儿童大脑型目前还有骨髓移植(Hematopoietic stem cell transplantation ,HSCT, including bone marrow transplant)能够治疗ALD,但成功率只有六成左右,另有三成七的机率是患者会产生排斥现象而导致死亡。HSCT具有死亡的风险,因此并不建议施行于已经有症状的ALD患者。脐带血(Umbilical cord blood)干细胞移植是对于那些ALD患者无法找到符合的HLA提供者的另一种治疗的选择方式,在年纪较小的病人身上会有比较好的预后。可是,即使骨髓移植成功,也不代表ALD就能被治愈,或是症状有减轻的现象。肾上腺脊髓性神经病变型则是至目前尚未有治疗方法(Beam et al.,2007)
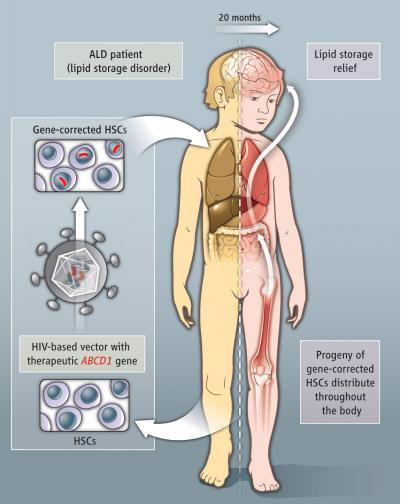
2009年11月在巴黎的INSERM (National Institute of Health and Research Medical)有一先驱研究的发表,研究者利用基因治疗结合血液干细胞治疗,对于ALD患者可能是一个有效的方式。巴黎的INSERM在两位病人身上,利用从患者身上取出造血干细胞(HSCs),使用基因治疗技术,也就是利用以AIDS病毒为载体,将带有正确ABCD1基因的病毒去感染患者自身的造血干细胞,替换原本因突变失去功能的ABCD1基因。最后再将已经替换好ABCD1基因的造血干细胞注射回患者身上治疗。正常的ALD蛋白大约在15%的血球细胞中会被表达出来,令人兴奋的是,即使是这样少量的蛋白也可以有效的减缓ALD对于脑部的伤害。病人实测的正常与非正常ABCD1基因细胞百分比大约是18%。这种方法可以减少因骨髓移植所带来的死亡与侵入性治疗的风险,并且也可以解决无适合骨髓提供者的问题(Cartier et al.,2009)。
资料来源:1.http://www.orpha.net
2.http://www.tfrd.org.tw/tfrd/intro_a#
3.百度百科
最后修改时间:2013年11月22日
