科学家们首次发现,在脊髓性肌萎缩症(spinal muscular atrophy,SMA)中受影响的控制肌肉运动的神经细胞或运动神经元,其线粒体存在缺陷,线粒体是为细胞供能的细胞器。运动神经元中线粒体结构和功能的受损出现于症状发生之前,提示其可能在疾病发展中发挥作用。
这些发现发表于《人类分子遗传学》(Human Molecular Genetics)期刊,为SMA的靶向疗法指出了新的可能性。
“恢复线粒体功能可能是一种新的SMA治疗策略”该研究资深作者Yongchao Ma博士说道。Ma博士是芝加哥安-罗伯特•H•卢里儿童医院斯坦利•曼勒儿童研究所的安•玛丽-弗朗西斯•克拉克医学博士研究学者,同时是美国西北大学芬伯格医学院儿科学、神经病学和生理学助理教授。
出生时罹患SMA的婴儿无法自行支撑头部或端坐,他们很少能活过2岁。
“虽然这种毁灭性疾病的遗传原因已经被识别,但我们的研究描述了线粒体功能障碍如何可能甚至在症状发生之前促进运动神经元破坏”Ma博士说,“我们的发现为SMA的发病机理提供了新的见解,这对于开发新疗法是至关重要的。”
在分析来自SMA小鼠和对照小鼠运动神经元的基因表达谱时,Ma博士和同事们首次发现线粒体与SMA有关。他们观察到,与许多线粒体功能相关的基因在SMA运动神经元中明显失调。
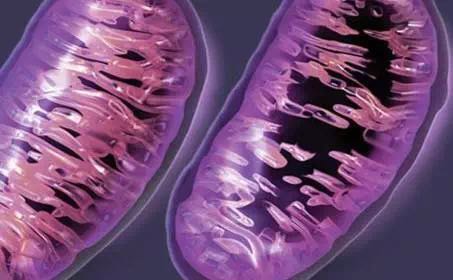
“这一发现是出乎意料的,促使我们在来自SMA小鼠模型的运动神经元中测试线粒体功能是否发生改变”Ma博士说道。
使用精细的技术,该研究发现,在SMA运动神经元中,线粒体以较慢的速度产生能量,耗尽神经能量。SMA线粒体的膜电位有所下降,这证明它不那么“健康”。它还具有升高的氧化应激水平,这对神经元是有毒的。线粒体的运动也遭受损害,导致其在神经和肌肉的接头处运动受阻,渗漏出毒素并最终扰乱神经肌肉连接。SMA运动神经元中的线粒体还会破碎和肿胀,这和该研究测得的线粒体功能缺陷相一致。
“运动神经元的能量需求较高,这使得它们对自身线粒体中的缺陷十分敏感”Ma博士说,“这些缺陷可能导致SMA中运动神经元变性退化的症状。”

该研究获得美国国立卫生研究院(NIH)、哈特韦尔基金会(HartwellFoundation)和怀特霍尔基金会(Whitehall Foundation)的支持。
芝加哥安-罗伯特•H•卢里儿童医院(Ann& Robert H. Lurie Children’s Hospital of Chicago)的研究通过斯坦利•曼勒儿童研究所(Stanley Manne Children’s Research Institute)进行。曼勒研究所关注于通过对科学知识的不懈追求来改善儿童健康,改变儿科医学,保障儿童更健康的未来。
卢里儿童医院被《美国新闻与世界报道》评选为美国顶级儿童医院之一。它还是西北大学芬伯格医学院(Northwestern University Feinberg School ofMedicine)的儿科训练基地。去年,全院共收治来自50个州和48个国家的超过174000名儿童患者。
原文链接:
--------------------
校审/曹文东、夏蓓
本文由中国罕见病网编译,转载请注明出处。

















