生活大爆炸第四季里有一集,Sheldon曾经跟Leonard探讨过一个问题,他怀疑自己有先天性巨结肠。很多好奇宝宝或者喜欢刨根问底的理科生可能都会想到一个问题:通常我们说的“遗传病”不是应该有家族史的吗?如果一个人的爸爸、妈妈都是正常人,为什么后代会得病呢?这种表面上根本看不出来“遗传”迹象和“遗传”来源的疾病,为什么会是遗传病呢?今天就努力给大家讲讲这个病的“前世今生”。

1888年一位丹麦儿科医生在世界上首次发现并且报道了先天性巨结肠的临床症状,这个病此后就用他的名字命名为:Hirschsprung’s disease(简称HSCR)。在1940年人们明确了疾病的病理改变以及上世纪50、60年代逐渐发展出不同手术方法来治疗、解决肠道梗阻的问题以来,先天性巨结肠的致死性变得不那么严重了。但是随之而来的,是很多“幸存”下来的患者有机会把他们携带的基因突变遗传给下一代,从而导致了越来越多的“家族性”患者的出现。
以此为契机,研究者很快于1993年定位出该病的第一个致病基因:RET,并陆续发现RET、EDNRB基因相互作用在肠神经系统发育中的重要作用以及RET基因内含子中的单核苷酸多态性变异可以显著增高患病风险等。诸多研究结果都证实:先天性巨结肠的发生与一个个体的“遗传背景”(genetic background)密不可分,“遗传”因素对于疾病发生的贡献大概可以达到80%左右。
OK,那么问题来了,为什么我们身边的HSCR患者属于家族性的并不多(即每个家庭只有一个患者),甚至有好多患者他们的父母都是正常(没有临床表现)的呢?这就要从HSCR这个疾病的复杂性和中国的特殊国情说起了。
首先,HSCR属于典型的人类复杂疾病,即使是疾病的主要基因RET,也只能解释约50%家族性HSCR患者和10-35%的散发病例的发病原因,并且RET基因参与HSCR的发生还有4个特性:
(1)编码区的罕见变异在整个基因范围内散在分布,不存在热点区域或热点位点,因而给拟诊或确诊患者的基因检测带来了困难;
(2)不同类型突变的外显率差异很大,家族内遗传方式常常不遵循经典的孟德尔遗传规律,不易建立明确的基因型-表型相关性,因此给遗传咨询乃至产前诊断的实施设置了几乎难以逾越的鸿沟;
(3)作用机制复杂-HSCR患者携带的突变多数都是功能失活突变并由此导致肠神经嵴细胞(Enteric Neural Crest Cell, ENCC)出现迁移、增殖或分化缺陷,而同一基因的功能获得突变却可引起IIA、IIB型多发性内分泌肿瘤或遗传性甲状腺髓样癌,既往文献报道及我们在临床工作中都遇到过家系内不同成员携带相同突变但是表型却有很大差异的实例;
(4)亚效等位基因的存在-内含子及上游非编码区内有多个单核苷酸多态性(single nucleotide polymorphism, SNP)位点,以增强子的角色参与基因表达水平调控,并能解释更多患者(尤其是最常见的短段型、男性患者)患病风险显著增加的原因。以上四条概括起来,大概可以通俗解释成三点:
一、患者父母可能临床表现较轻,因此没有就医、确诊为HSCR;
二、患者的主要致病突变有可能确实遗传自父母一方,但由于同一突变在不同个体的“外显”程度(可以简单理解成表现度)不同,有可能被忽视;
三、RET基因的功能除了受一些罕见突变的影响外,还有很多其他调控因素,患者可能从父母双方分别获得了风险变异位点,两者或多者“叠加”最终导致了疾病的发生。其次,中国的独生子女政策可能在某种程度上大大降低或者切断了家族性患者的出现概率,因此我们推测,随着“全面二孩”时代的来临,或许3-5年内就可以观察到先天性巨结肠的一个“井喷”现象。
目前分子遗传学家的共识是:RET基因功能异常是HSCR发生的必要条件,几乎每个患者,都一定有这个基因的功能下降,原因可能是罕见突变,也可能是SNP导致的基因转录水平受损。今天先来说说前者:
依据美国医学遗传学与基因组学学会(The American Collegeof Medical Genetics and Genomics, ACMG)制定的序列变异解读指南,我们可以对RET基因上发现的罕见突变的致病性进行大致划分如下:
(1)致病性变异-证据等级:非常强:先天性巨结肠(HSCR)的主要发病机制为RET基因及其编码蛋白的功能缺失(loss-of-function, LOF),在此情况下,下列几种无功能变异应被视为证据非常强的致病性变异:无义突变、移码突变、经典±1或±2的剪接突变、起始密码子变异、单个或多个外显子缺失。
(2)致病性变异-证据等级:强:包括与已知的致病性变异有相同的氨基酸改变(错义突变)、患者自身的新发变异且无家族史、体内外功能实验已明确会导致基因功能受损的变异、变异出现在HSCR群体中的频率显著高于对照群体等。
(3)致病性变异-证据等级:中等:包括:位于关键蛋白功能域的变异、数据库显示正常人未发现的变异(或极低频位点)、在非重复区内的非移码插入/缺失、终止密码子丢失、已知致病位点发生的未报到过的错义突变、由于父母一方或双方DNA样本缺失导致的无法验证的新发变异等。
(4)致病性变异-证据等级:支持:包括:在家系内的多个患者都可检测到的变异、多种统计方法均预测会对基因或基因产物造成有害影响的变异等。
(5)文献报道过的可能致病变异举例:
有些文献曾经报道过很多在HSCR患者检查出来的突变(或变异),这些突变(或变异)如果有进行过相应的功能学实验并且证实可以影响RET编码蛋白质的功能,它的致病性就基本可以确立;如果是没有功能实验但是在多个不同种族或者同种族不相关的患者内被检出过,致病的可能性也是比较大的。所以我们在评判一个变异的致病性时,通常要考虑很多方面的因素,而且有些情况下确实没办法给出一个100%确定的结论,这是复杂疾病研究的共性。
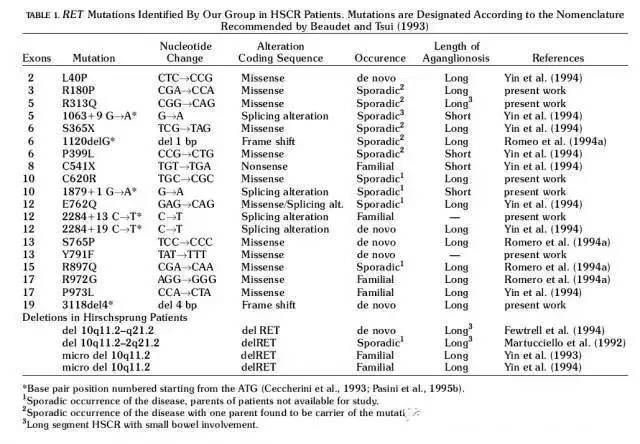
摘自参考文献:Hum Mutat. 1997, 9 (3): 243-249, SeriM et al (Frequency of RET mutations in long- and short-segment Hirschsprungdisease)
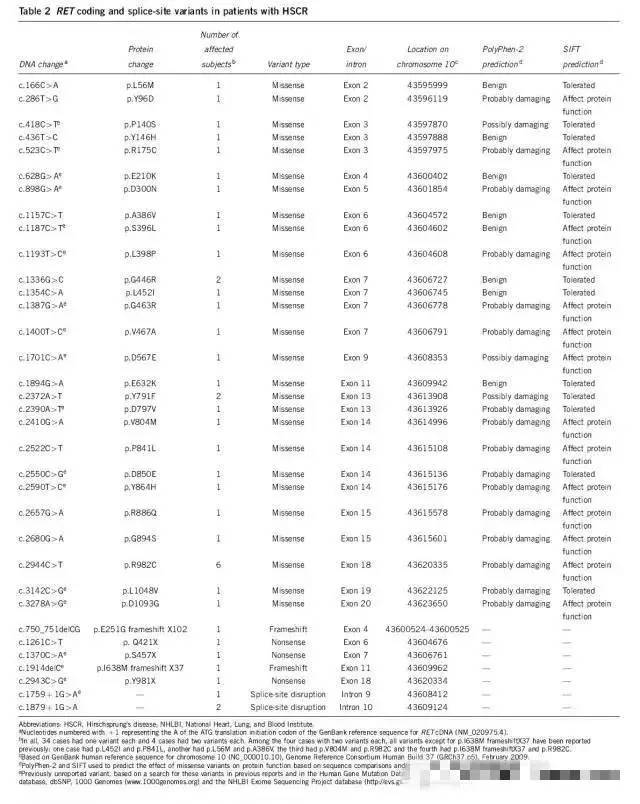
摘自参考文献:J Hum Genet. 2012,57 (8): 485-493,Carter TC et al. Hirschsprung's disease and variants in genes that regulateenteric neural crest cell proliferation, migration and differentiation.
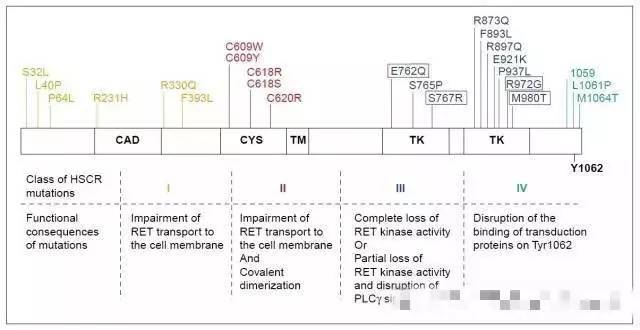
摘自参考文献:Trends Genet. 2001, 17 (10): 580-589,Manié S et al. The RET receptor: function in development and dysfunction incongenital malformation. 框内突变部分降低RET激酶活性丧失。
总的规律:
①胞外段的突变可能影响RET蛋白质成熟和细胞膜表面的表达;
②半胱氨酸富集区的突变可能会影响蛋白质二聚体的共价结合和转运;
③酪氨酸激酶功能区的突变可能会改变酶的催化活性或影响蛋白质的结构稳定;
④胞内段的突变可能会影响受体与细胞内信号蛋白的结合。
文末划重点:
(1)如果你的主治大夫告诉你先天性巨结肠是不遗传的,再生育时可以完全不用考虑再发风险,那么你可能已经开启了一段“无知者无畏”的旅程;
(2)对于严重类型的HSCR患者,即长段型和全结肠受累型,我们强烈建议家长给孩子进行相关基因检测,因为发现致病基因突变的几率相对较大,而明确的分子遗传学诊断是遗传咨询的必要、先决条件。

















