5月24日,美国FDA批准了诺华公司的第一个用于治疗2岁以下脊髓性肌萎缩症(spinal muscular atrophy,SMA)的基因疗法——Zolgensma。这个基因疗法的获批标志着用基因和细胞疗法 (cell and gene therapy) 来治疗疾病的一个里程碑。
(图片来源:https://www.theadvocate.com.au)
该基因疗法主要针对一型脊髓性肌萎缩症(SMA1)——最严重的一种类型——大部分患有这种病的婴儿最后会因为呼吸衰竭而活不过2岁。
这项药物的成功研发和患者、家属、科研领域和制药公司的共同努力不可分割。本文就来讲讲脊髓性肌萎缩症药物研发背后的故事:一对父母是怎样为了治疗患绝症的女儿,从而推动了整个药物研发的进步!
Arya Singh出生于2000年3月。起初,她的发育进程和书本上写的差不多,在正常的时间会坐会爬。但是到了学走路的时候,她就停滞不前了,17个月大的时候还是走四五步就会跌倒。她的儿科医生安慰她父母说,这孩子可能发育比别的孩子晚一些。
2001年8月,Arya的父母带着她去参加一个派对。派对上的一位医生朋友看到她走路磕磕绊绊的样子,觉得很不对劲,建议立刻去看神经科医生。
于是,他们找了好几个专科医生,做了各种检查,能够确诊的血检要一个多月才能出结果。这个等待过程对Arya的妈妈Loren Eng——一位正怀着8个月的老二的美籍华裔——来说,显得尤其漫长焦灼。就在Loren预产期前两天,神经科医生打电话告诉他们, Arya患有脊髓性肌萎缩症(Spinal muscular atrophy,简称SMA)。Loren还没来得及反应,医生就快速说了一句“我非常抱歉”,挂断了电话。
什么是SMA?
脊髓性肌萎缩症,简称SMA,是一种常染色体隐性遗传的神经退行性疾病,是由人体内2个拷贝(分别来自于父母)的SMN1基因都变异/缺失造成的。SMN1是对运动神经元的存活至关重要的基因,它的缺失会导致控制肌肉的神经逐渐退化,肌肉萎缩,最终大部分会因为呼吸肌衰竭而死亡。很多患有SMA的婴儿在出生几周或者几个月内看着也许和正常的孩子一样,但是他们的神经和肌肉会很快衰退,大部分孩子活不过2岁。
据统计,每8000个出生的婴幼儿就有一个患这种疾病,每50个人中就有一个是突变基因的携带者[1]。
如果父母双方都是SMN1基因变异的携带者(一个拷贝正常,另一个拷贝变异/缺失),那么生出的孩子有25%的几率得SMA。
SMA的病理
1891年,奥地利神经病学家Guido Werdnig首先描述了最严重的I型SMA的症状。可是它的遗传起因一直不详。直到1995年,法国研究人员在5号染色体上发现了一个基因, 叫做SMN1。
SMN1基因突变或者缺失,是造成SMA的主要原因。
几乎所有的人,包括SMA患者,都有一个和SMN1几乎一样的基因叫SMN2。SMN2在基因序列上与SMN1几乎相同,只有5个核苷酸不一样。但是,就在这5个不同的核苷酸中,有一个是在外显子7上面的C到T的变化。这会导致转录剪接SMN2基因的时候,90%会跳过外显子7,产生截短的、无功能并且被快速降解的不稳定蛋白。
只有大约10%的SMN2转录会产生全长蛋白,为SMA患者提供少量的SMN蛋白以维持脊髓运动神经元的存活。
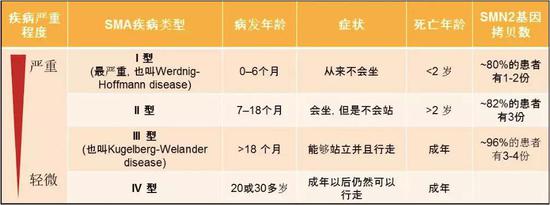
在SMA患者体内,SMN2基因拷贝数有高有低。拷贝数越高,症状越轻。超过95%的SMA患者至少保留1份SMN2基因[2]。但是,虽然SMN2基因拷贝数与疾病严重程度有关,但并不能准确预测疾病的严重程度,因为有些研究发现可能还有别的因素可以影响症状。SMA根据症状的严重程度分为四型。下面的表格就归纳了四型的区别。
Arya的父母在接下来的24小时里面慌忙安排血检,看马上要出生的老二是不是也会得这种病(好消息是他没有)。一些医生安慰他们说,Arya的症状相对温和,应该可以活到成年。但是随着年龄的增长,病情会逐渐恶化,她会失去行动能力,向残疾靠拢。
疯狂给Arya寻找治疗方案的过程中,她的父母发现了一个令人震惊的事实:针对SMA的药物研发当时少得可怜。美国国立卫生研究院每年拨给SMA的研究资金是1300万美元, 是花在囊肿性纤维化(cystic fibrosis,也是一种遗传疾病)的九分之一。生物制药公司也大都忽视了这种疾病。
美国每年用于医学研究的经费大约是280亿美元。即使这样,有好些致命的疾病还是被遗忘了。它们没有名人代言,有些还有着奇怪的不太好读的名字,或者是病人死去得太快,没有足够活着的人来向公众宣传。
一个最好的例子就是胰腺癌。美国政府对胰腺癌研究的资金投入只有前列腺癌的十分之一,政府对后者的重视很大程度得益于名人效应——虽然这两种癌症的患者人数差不多,但是前列腺癌有很多出名的幸存者,比如著名的自行车赛职业车手兰斯·阿姆斯特朗和前纽约市长鲁迪·朱利安尼。

鲁迪·朱利安尼被《时代》杂志列为2001年风云人物
很不幸,这些问题SMA都有。
首先,对于大型制药公司而言, SMA病人数量太少了,还不到囊肿性纤维化病人数量的一半。投入的研究和临床试验的成本并不能得到足够的经济回报。
其次,不同类型的SMA传统上被冠上各种不起眼的名字,诸如“Werdnig-Hoffman病”和“Kugelberg-Welander综合症”,还经常和肌肉萎缩症(Muscular dystrophy)相混淆。 直到最近十年,它才被系统地划分。
然而,Arya的父母也不是普通人。Arya的爸爸,Dinakar Singh ,是华尔街著名的人物。他当时是高盛的股票交易师和合伙人,现在是对冲基金TPG-Axon Capital的创始人,管理着28亿美元的资产。Arya的妈妈,Loren Eng,曾经在投资银行工作,是斯坦福大学教育和经济双硕士。他们自己拿出了1500万美元建立了一个叫SMA Foundation的慈善机构,致力资助这项疾病的研究。Loren自己出任基金会主席。这个基金会的目标是:趁还来得及的时候,尽快给Arya和其他25,000名患有SMA的孩子找到治疗方法。
他们具体是怎么做的呢?
1争取政府的支持
其实在2002年初,美国国立卫生研究院的神经科学家Kenneth Fischbeck就已经提出了一项创新项目来开发针对SMA的新药。但是,由于政府机构的重重官僚主义,到了2003年初,这个计划还是拖拖拉拉的没有推行下去。
Loren找到了50位科学家,其中包括DNA发现者詹姆斯∙沃森,向美国国立卫生研究院请愿更多的SMA研究经费。她还雇了一名说客,把各种SMA患者团体组织起来,给国会写信,晓之以理,动之以情。2003年初,他们就发出了400多封邮件。Loren和Dinakar还去和参议员见面游说。
功夫不负有心人,2003年4月的一次国会听证会上,一位参议员质问美国国立卫生研究院的负责人为什么还没有推行Kenneth Fischbeck的SMA创新药计划。6个月后,该计划正式启动,资金为2.22千万美元。
2游说生物制药公司
2004年春天,Dinakar和Loren决定弄一个像投资银行的巡回展,来吸引生物制药公司的兴趣。 他们准备了PPT文档,估算了SMA药物的潜在年销售额(2.5 ~7.5亿美元),以及这种新药可能有的其它衍生用途。最大的卖点是,相对于别的疾病,SMA的病理已经研究得比较透彻,很快找到治疗方案的机会很大。他们也愿意给有意向的公司提供一笔可观的研究经费。
可是,实际操作还是比想像的要困难得多。2004年5月的一个神经生物学年会上,他们邀请了七家制药公司的老总吃晚饭, 最后只来了3个。
有些公司的高管们担心不研究肥胖这样的热门领域,反而转向罕见病的研究会让公司股票贬值。还有一些公司的顾虑是传统上儿科药物的临床试验很难做,也很难得到美国食品和药物管理局的批准。还有一些人指出,测试药物所必需的小鼠模型和其他关键资源分散在各个大学实验室中,许多实验室要求一笔可观的费用才同意共享这些资源。
3促进学术界和工业界的合作
Loren亲自飞到台湾,说服那儿的一个实验室把 SMA小鼠模型和别人分享, 并且多次主办研讨会,邀请学术界和工业界著名的科学家来做讲座和相互交流。
最值得一提的是,和其他一些病人倡导组织不同,他们不是给研究人员一笔钱就不管了,而是积极参与其中,聘请专家,推动研究人员合作而不是互相竞争,尽一切可能给研究提供方便。当年, Arya的一些组织被送来笔者的实验室做一些生物分析。当我们表示最好有一个健康人来做对照的时候,Loren二话不说,立刻送来了她自己的样品。
所有这些都是在和时间赛跑,因为2003年底,Arya已经失去了控制脖子上肌肉的能力。
在他们不懈的努力之下,终于有很多公司开始了SMA的药物研发。
例如,麻省剑桥市的公司Curis拿到了来自SMA Foundation的三年540万美元的资金,利用其独特的在试管中培养运动神经元的技术来筛选可能的药物。
2016年底,由百建公司(Biogen)和 Ionis Pharmaceuticals (伊奥尼斯制药)联合开发的第一个SMA药物Spinraza被批准上市。
Loren和Dinakar在Spinraza的研发过程中也起到了关键的作用。
1999年,一位名叫Adrian Krainer的科学家开始研究如何可以通过调整SMN2的蛋白水平来缓解SMA的症状。几乎在同一时间,Ionis Pharmaceuticals 也正在寻找机会来测试他们可以微调蛋白质翻译的新技术。他们决定合作,用Ionis的技术来微调SMN2的蛋白水平,并且取得了很快的进展。到了2004年,他们遇到了一个问题:能够用来测试药物效应的小鼠模型在大半个地球以外的台湾的一个实验室。
Loren亲自飞去台湾,把一批小鼠运来了美国。随之而来的测试结果令人鼓舞:这些小鼠从仅能存活10天到用完药物以后很多能存活超过一年。
但是,像Ionis这样的小型生物技术公司没有足够的资源来完成人体临床试验,他们需要和一个大制药公司合作。于是,在2011年夏天的SMA基金会晚宴上, Loren安排Ionis的高管坐在百建公司的首席医疗官旁边,最终促进了这两个公司的联手。
Spinraza的缺点是需要脊髓腔内注射,每个月需要打一针。而且很贵,每个病人第一年的费用大约是75万美元,随后是每年37万5千美元。
这次被批准上市的Zolgensma比起Spinraza又改善了很多。它只需要静脉注射一针,目前的数据看至少2年都有很好的效果。但是,它还是很贵,5年每年支付42万5美金,是有史以来单价最贵的药物。
还有更多的药物正在各期临床试验中,其中很多是可以口服的药片。
虽然从5岁开始,Arya就不得不坐轮椅,她的手臂上几乎没有肌肉,儿科医生打针只能打在腿上,但现在,她已经是加州某著名大学的大学生。

Arya和父母
下面录像里名叫Oskar来自波兰的男娃,三个月大的时候被诊断出患有最严重的I型SMA,一般活不过两岁的生日。他加入了诺华公司正在开发的可以口服的SMA药物LMI070的临床试验,2个月以后学会了翻身,2年以后可以很好地抓玩具。
相信在不久的将来,这种疾病就会有更多的疗法!对于一个做药的人,能够看到一个像Oskar一样的患者因为自己的努力而受益,也就觉得所有的努力都是值得的。

















