脊髓性肌萎缩症(SMA)是一种罕见的常染色体隐性遗传病,在新生儿中的发病率约为1/10000[1]。
SMA的病因明确,由于5号染色体长臂SMN1基因的7号外显子纯合缺失或突变,SMA患者无法产生足够水平的运动神经元存活(SMN)蛋白,最终导致运动神经元进行性退化。
SMA的患病人群从新生儿到成年人,病发症状从行走障碍到无法独坐,严重的连吞咽、咀嚼、甚至呼吸功能都逐渐退化。有统计数据表明,SMA是导致2岁以下儿童死亡的头号遗传病杀手[2]。
近年来的研究发现,在5号染色体上还有一个SMN2基因,它与SMN1基因高度同源,二者均能表达SMN蛋白。不过,SMN2上的c.840C>T变异,会导致90%的SMN2的前信使RNA的转录产物发生选择性剪接,仅有10%的SMN蛋白具有功能[3]。
由于不同患者的SMN2基因拷贝数存在差异,科学家已经发现SMN2拷贝数与SMA的表型严重程度相关。理论上讲,SMN2基因的拷贝数越多,表达出的功能性SMN蛋白的量就越多,患者生存时间越长、症状越轻[4]。
基于以上机制,经过科学家的不懈努力,自2016年以来,已有3种药物获得美国FDA批准:Nusinersen(诺西那生钠)、Risdiplam(利司扑兰)和Onasemnogene Abeparvovec-xioi(后文简称OA),其中前两种药物分别于2019年和2021年在国内上市。
从作用机制上看,诺西那生钠和利司扑兰都是通过改变SMN2剪接、上调SMN蛋白表达水平而发挥治疗作用的,患者需要终生用药;而OA则是一种基因疗法,通过腺病毒把目标基因转染进入组织(但一般不会整合到宿主基因组)起效[5]。
之前有研究表明,在患儿出现症状前使用诺西那生钠治疗的临床结局,优于出现症状后治疗的患儿[6]。那么更早地实施基因疗法,是否也会进一步改善患儿的临床结局呢?
前不久,由美国特殊儿童特诊所(Clinic for Special Children)Kevin A. Strauss领衔的研究团队,在著名期刊《自然·医学》杂志发表了两项III期临床试验数据[7,8],提示了SMA的早期基因治疗对患儿预后效果的积极影响。
Strauss团队发现,对患儿实施药物治疗的起始时间越早,患儿神经肌肉退行性病变的严重程度越轻,同时,患儿的SMN2拷贝数越多,药物治疗的效果越好。而从给药方案来看,OA具有单剂量、一次性的潜在优势。

论文首页截图
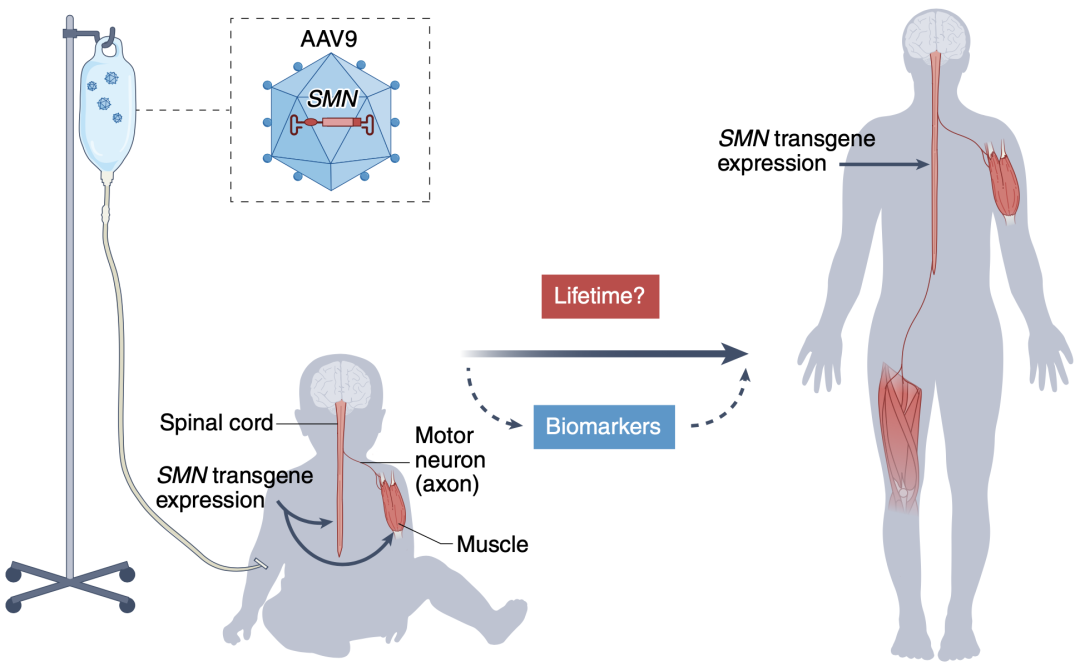
与改变SMN2剪接的治疗机制不同,OA是通过一种9型自身互补型腺相关病毒(AAV9)的转染,将目的基因SMN运送入靶组织,而对疾病发挥治疗作用的。
(注:AAV是一种非致病性病毒,能够在不整合到宿主基因组的情况下实现持续的转基因表达,并且很少激活机体的免疫反应。AAV有几种血清型,不同的血清型AAV的组织亲嗜性不同,其中AAV9对中枢神经系统和肌肉组织有亲嗜性,就被选为SMA的基因治疗载体。)
基于这种作用机制,OA被一次性静脉注射进入体内后,就可以穿透血脑屏障、转染神经元细胞和神经胶质细胞,也能转染肌肉、肝脏和其他外周组织,在转染组织中表达SMN蛋白,快速和持续地改善SMA患者的运动功能。而且,多项针对SMA患儿临床试验结果以及药物上市后的数据[9-12],都证实了OA对SMA患者的治疗作用。
但是,前期进行的临床试验,多是面向已经出现症状的SMA患儿的,而OA对未发病患儿治疗的有效性和安全性如何,治疗时机和预测分型对治疗效果的影响如何是未知的。
为了回答上述问题,Strauss等人开展了一项纳入29名已经确诊,但暂未出现临床症状的SMA患儿的临床研究(SPR1NT),其中14名有2个SMN2拷贝(平均年龄21天,下文简称两拷贝),15名有3个SMN2拷贝(中位年龄32天,下文简称三拷贝)。
研究人员对入组患儿进行了单剂量的OA静脉注射治疗,并在治疗后开展了为期18-24个月的随访,随访内容包括:30秒独坐,至少3秒独立站立,14个月大时的无事件生存率等。
随访结果显示,在症状发生前接受OA治疗,两拷贝患儿中79%可以实现独自站立、64%可以实现独立行走,而三拷贝患儿可以独自行走的人数比例更高达93%。相比之下,相同基因型的两拷贝患儿在症状发生后接受OA治疗,中位年龄4个月时能够独立站立或行走的患儿只有3-5%[7,8]。
值得一提的是,该项研究中的所有患儿,均在治疗后达到了Bayley-III幼儿发育量表(BSID)和世卫组织多中心生长参考研究(WHO-MGRS)定义的运动发育里程碑,并在24个月随访结束时保持这一里程碑。
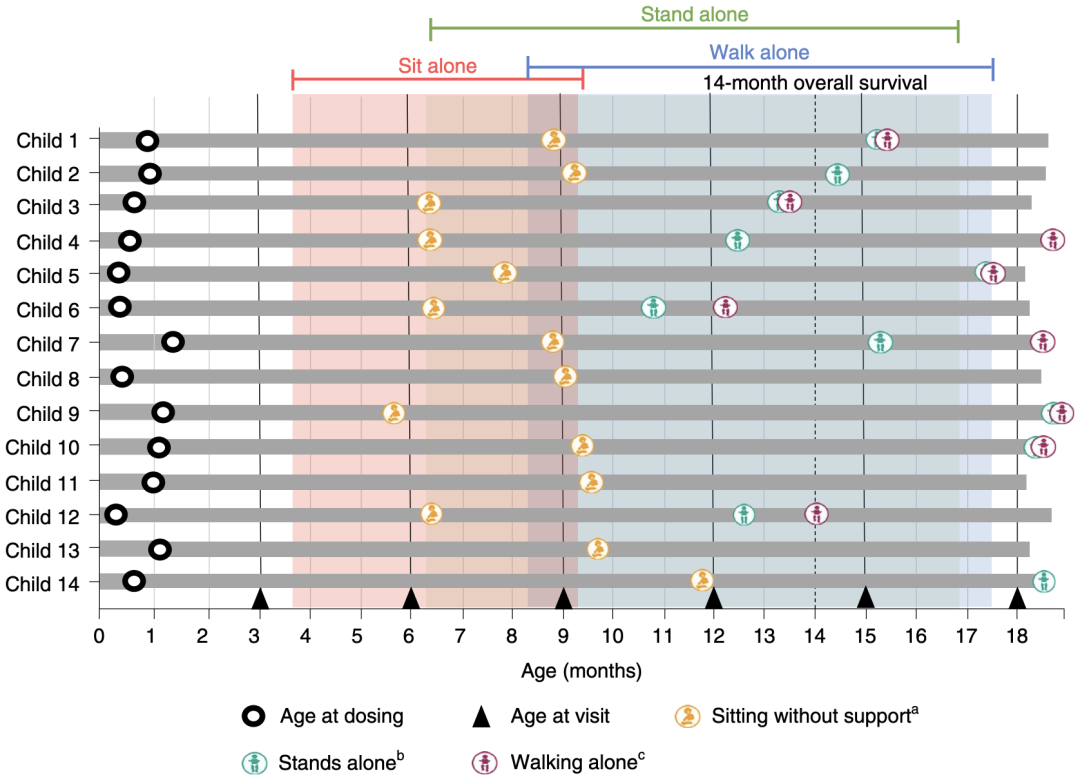
SMN2两拷贝患儿经OA治疗后的运动发育里程碑
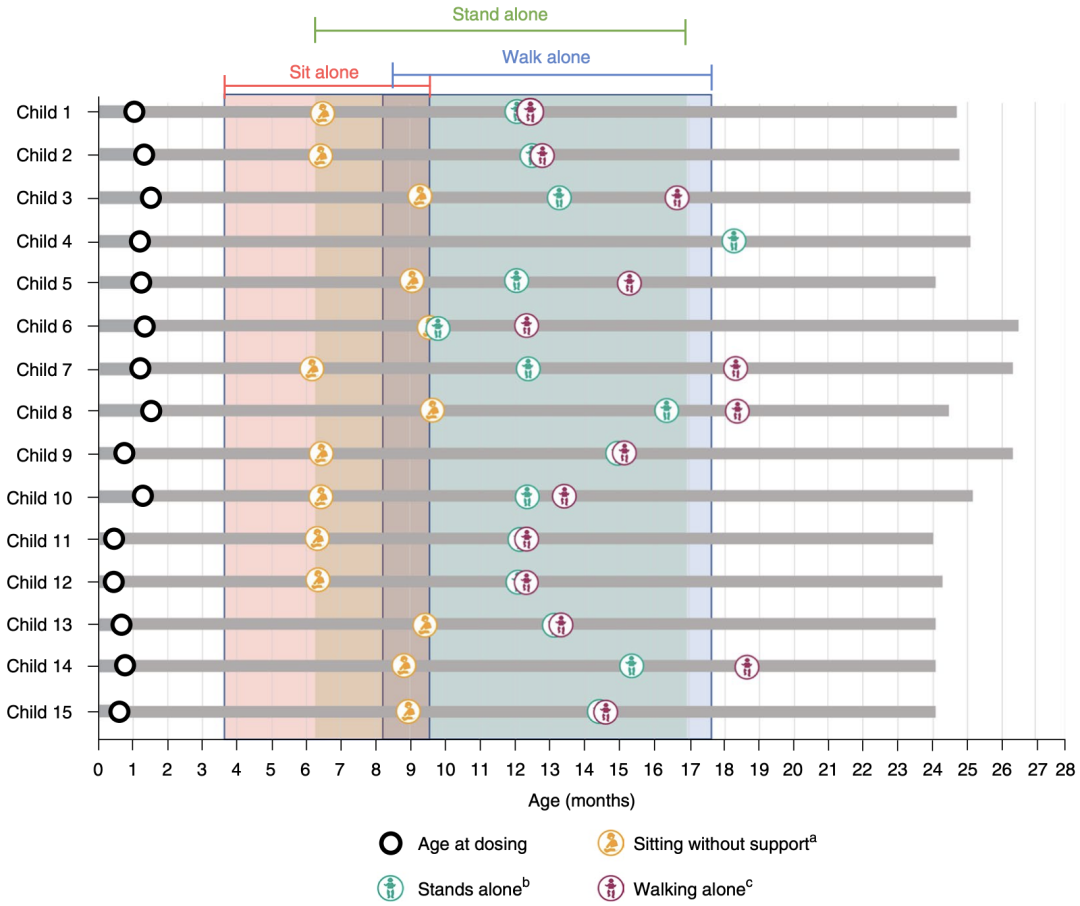
SMN2三拷贝患儿经OA治疗后的运动发育里程碑
这项研究除了评估入组患儿在接受治疗后的独坐、独站和独走的能力这些主要研究终点外,还对他们的无永久通气存活时间和无呼吸机存活率进行了统计。
研究结果显示,无论是两拷贝还是三拷贝的患儿,都达到了14个月的无永久通气存活时间,且24个月的无呼吸机存活率达到100%。
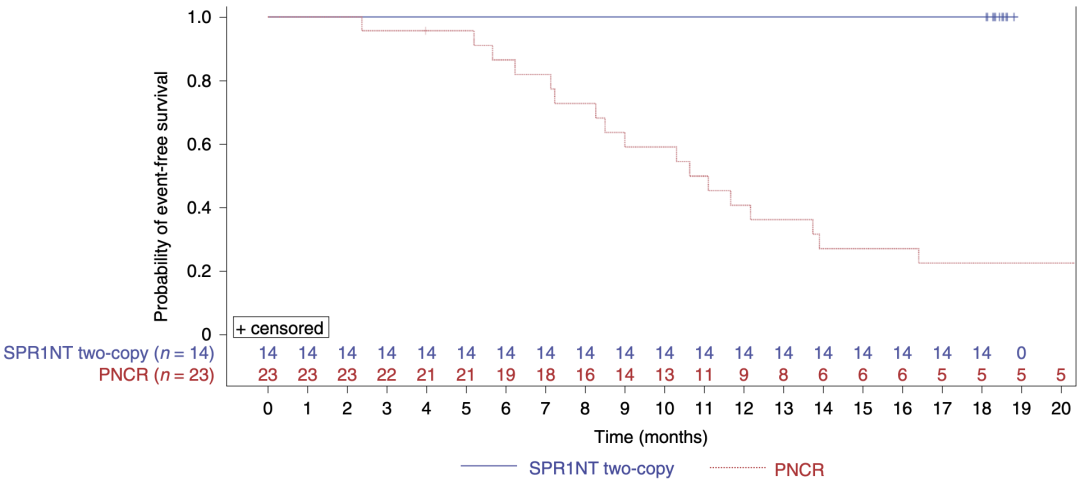
与另一项临床研究(PNCR)中的未治疗队列的无事件生存期的对比
虽然,OA的治疗大大改善了SMA患儿的症状,但在治疗过程中已经产生或可能产生的毒性问题,也不能被忽视。
比如,为了减轻患儿对AAV9的免疫反应,所有入组患儿均在OA注射前一天开始口服泼尼松龙,并至少服用至OA注射后30天。这导致了多数患儿都出现过治疗相关不良事件:肝毒性、血小板减少、心脏毒性、血栓性微血管病和背根神经节病变。
总体来看,无论是OA治疗的这项临床研究结果,还是此前已经完成的诺西那生钠的临床试验结果[6],都显示了症状前治疗的患儿的结局是优于症状后治疗的患儿的(利司扑兰治疗症状前患儿的临床试验[NCT03779334]正在进行中)。这也提示我们,治疗时机的选择比治疗方案的选择可能更重要。
救治SMA患儿,是一场与时间的赛跑,及早筛查出患儿并立即开始药物治疗,可以有效改善患儿生存发育的结局,挽救生命。
参考文献:
1.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27-32. doi:10.1038/ejhg.2011.134
2.Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. Natl Vital Stat Rep. 2010;59(2):1-52.
3.Arnold ES, Fischbeck KH. Spinal muscular atrophy. Handb Clin Neurol. 2018;148:591-601. doi:10.1016/B978-0-444-64076-5.00038-7
4.Serra-Juhe C, Tizzano EF. Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors. Eur J Hum Genet. 2019;27(12):1774-1782. doi:10.1038/s41431-019-0415-4
5.Nicolau S, Waldrop MA, Connolly AM, Mendell JR. Spinal Muscular Atrophy. Semin Pediatr Neurol. 2021;37:100878. doi:10.1016/j.spen.2021.100878
6.De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842-856. doi:10.1016/j.nmd.2019.09.007
7.Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat Med. 2022;28(7):1381-1389. doi:10.1038/s41591-022-01866-4
8.Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med. 2022;28(7):1390-1397. doi:10.1038/s41591-022-01867-3
9.Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723-1732. doi:10.1056/NEJMoa1702752
10.De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842-856. doi:10.1016/j.nmd.2019.09.007
11.Darras BT, Masson R, Mazurkiewicz-Bełdzińska M, et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N Engl J Med. 2021;385(5):427-435. doi:10.1056/NEJMoa2102047
12.Markati T, Fisher G, Ramdas S, Servais L. Risdiplam: an investigational survival motor neuron 2 (SMN2) splicing modifier for spinal muscular atrophy (SMA). Expert Opin Investig Drugs. 2022;31(5):451-461. doi:10.1080/13543784.2022.2056836

















