肌萎缩侧索硬化(ALS)是一种进展性的神经退行性疾病,涉及上下运动神经元的丢失,大多数病例在发病3-5年内死亡。目前为止,尚未发现能够阻止或逆转疾病进展的治疗方法或药物。利鲁唑是一种神经保护剂,虽然治疗效果有限,但长期以来仍然是ALS的疾病修正治疗药物。统计学上来看,依达拉奉显示出显著减缓ALS疾病进展的作用。苯丁酸钠-牛磺酸二醇(PB-TURSO)于2022年在加拿大获批,具有显著减缓疾病进展并延长生存期的临床效果。此外,大多数临床试验都集中在细胞通路的小分子,靶向谷氨酸能、凋亡、炎症和氧化应激等假说机制。最近,干细胞移植和利用其他生物制剂进行的临床试验已显示初步临床数据和效果。本篇盘点ALS的现有治疗药物和目前研发中的治疗方法[1]。
已获批的ALS治疗药物
1. 利鲁唑
利鲁唑是一种神经保护剂,被认为具有抗谷氨酸能活性,它是已获批准疾病修正疗法之一,可适度提高ALS患者的存活率。研究表明,利鲁唑通过增加谷氨酸重摄取、抑制谷氨酸受体和阻断钠通道共同减少了兴奋性毒性,从而降低了神经元死亡(见图1)。此外,利鲁唑还通过刺激脑源性神经营养因子及调节谷氨酸从而影响神经可塑性。
早年的一项里程碑式的研究发表于新英格兰医学杂志[2],这是一项前瞻、双盲、安慰剂对照试验,纳入155例肌萎缩侧索硬化患者,随机接受利鲁唑(100mg/天)和安慰剂。经过12个月的随访,发现利鲁唑有效提高患者生存率(74% VS 58%,P=0.014)。并且利鲁唑组患者肌力的恶化明显慢于安慰剂组(P = 0.028)。
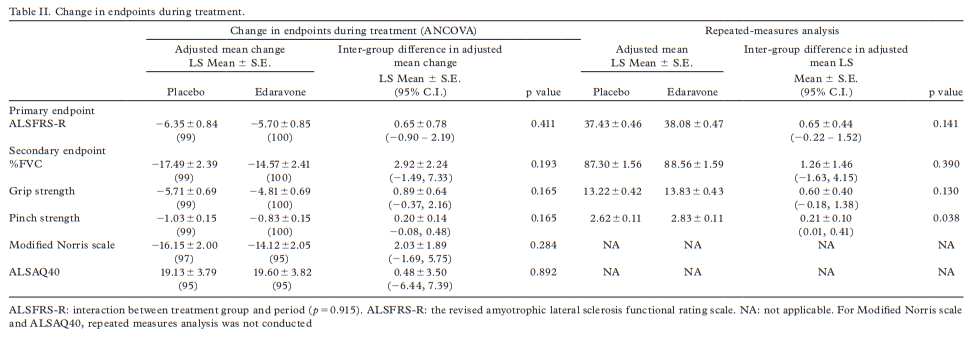
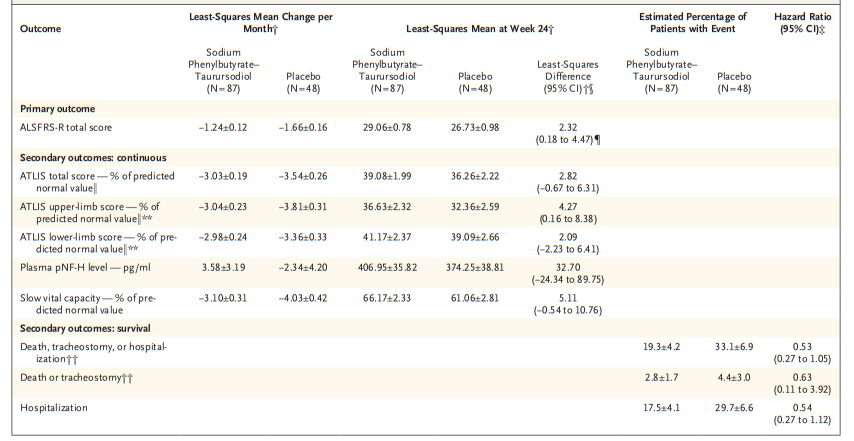
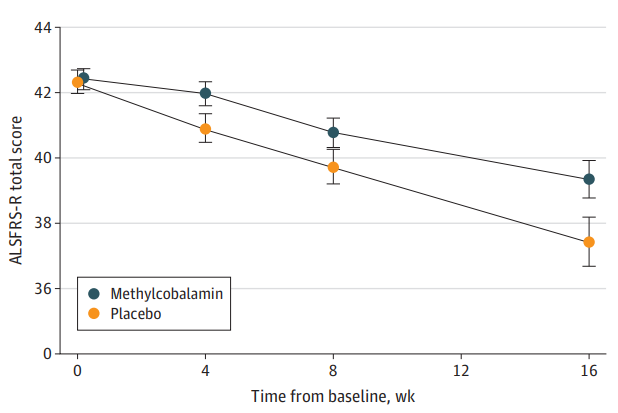
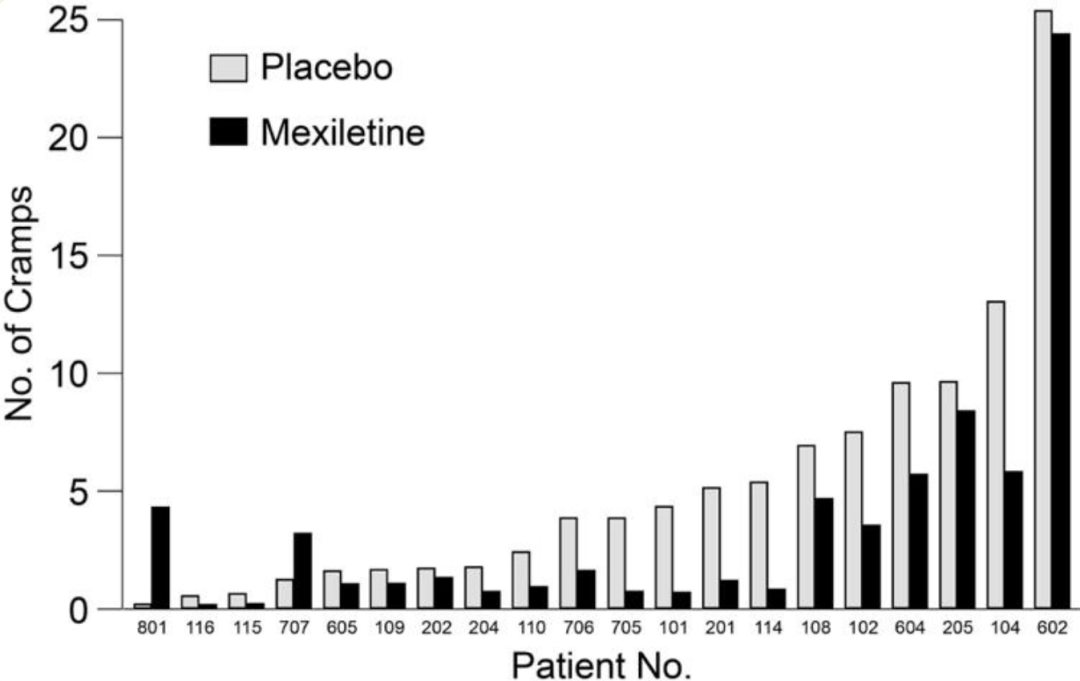
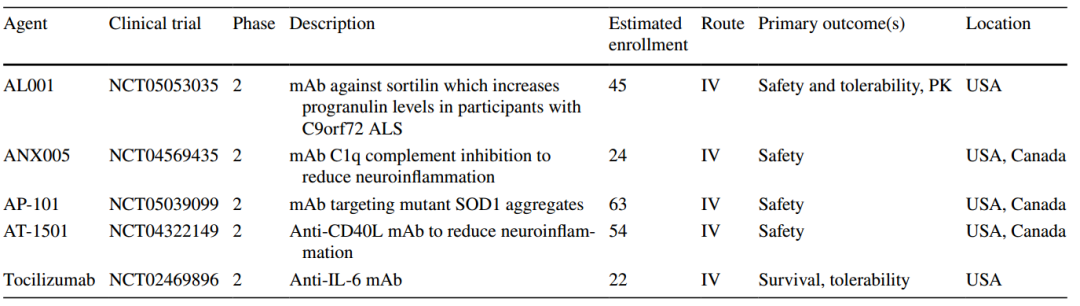
图1 利鲁唑发挥ALS治疗作用假说图 (突触前和突触后神经元(蓝色),神经递质穿过突触(黄色)和星形胶质细胞(紫色),NMDA受体(红色)、钠通道(蓝色)、EAAT2通道(紫色)) 在接下来发表在柳叶刀杂志上最佳给药剂量的研究中[3],结果显示在18个月随访期后,安慰剂、50mg、100mg、200mg利鲁唑组患者生存率分别为50.4%、55.3%、56.8%和57.8%。其中200mg利鲁唑组不良反应发生率最高。100mg利鲁唑具有最佳风险获益比。 并且,对人群数据进行回顾,发现利鲁唑在现实世界中可能使患者获得更长的生存延长期,中位生存期延长6-19个月[4]。 给药形式包括口服片剂、混悬剂和舌下崩解制剂,三者具有相同生物等效性。考虑到大多数(高达85%)的ALS患者在其病程的某个时间点会出现吞咽困难的症状,片剂给药实现较为困难。虽然药片可以被碾碎,但会造成给药剂量不准确,此时上述替代给药途径可以解决此类问题。利鲁唑一般耐受性良好,副作用包括疲劳、肝毒性和胃肠道症状[2, 3],应定期进行肝功能检查,监测肝毒性。 2. 依达拉奉 依达拉奉最初被批准用于中风,确切的作用机制尚不清楚。但体外和体内研究表明,依达拉奉可能通过清除自由基和抑制神经炎症反应而发挥保护作用。目前的临床研究的结论尚未统一。 一项来自日本的双盲、平行组设计、安慰剂对照的临床研究[5],纳入206例患者,静脉予以依达拉奉,以ALS功能评分量表(ALSFRS-R)评价患者。安慰剂组在24周治疗期间ALSFRS-R评分的变化为-6.35±0.84,依达拉奉组为-5.70±0.85。结果显示依达拉奉组ALSFRS-R评分变化略优于安慰剂组,但两组无统计学差异,依达拉奉的临床疗效似乎尚未得到证实(见图2)。但在亚组分析中发现 ,对于ALS分级在1级或2级,用力肺活量达到80%以上,且病程小于2年的患者中,应用依达拉奉ALSFRS-R评分的变化为-5.01±0.64,安慰剂组为-7.50±0.66,两者具有显著统计学差异,说明对于该亚组患者依达拉奉可有效减缓神经功能衰退。 图2 依达拉奉和安慰剂对ALSFRS-R评分的影响 最近,一项来自德国多中心、评分匹配的队列研究用以评价静脉依达拉奉在真实世界中对ALS患者的长期安全性和有效性[7]。最终分析324名患者,其中194名患者联合应用依达拉奉和利鲁唑,130名患者接受标准治疗,即单独应用利鲁唑。结果显示,使用依达拉奉治疗的患者的疾病进展与接受标准治疗(ALSFRS-R评分-0.91 VS -0.85)的患者没有差异。在生存率、通气时间和疾病进展变化的次要终点方面同样没有观察到显着差异。该研究表明同单独使用标准治疗相比,静脉给予依达拉奉可能无法提供临床相关的额外获益。 依达拉奉在临床试验中耐受性良好,最常见的副作用包括淤青(15%)、步态障碍(13%)和头痛(10%)[6]。 自2015年以来,依达拉奉已在日本和韩国被批准用于的肌萎缩侧索硬化,美国、加拿大、瑞士、中国、印度尼西亚、马来西亚和泰国获批紧随其后,2022年5月,依达拉奉口服制剂在美国获批,口服制剂的出现为运动功能残疾的病人带来极大地便利性。在美国,最近的数据表明,大约有13%-25%的ALS患者在接受静脉依达拉奉,而且鉴于口服药物的便利性,在未来依达拉奉的使用率可能会进一步增加。 3. 苯丁酸钠-牛磺酸二醇(PB-Turso;又称AMX0035) AMX0035是一种固定剂量的苯丁酸钠和牛磺酸二醇(PB-Turso)的联合制剂。苯丁酸钠以往被用来治疗尿素循环障碍中的高氨血症,牛磺酸二醇用于治疗慢性胆汁淤积性肝病。苯丁酸钠和牛磺酸二醇被认为可以减少内质网应激和线粒体功能障碍,减少ALS中神经细胞死亡,延长患者生存时限。 近年发表于新英格兰医学杂志的多中心、随机、双盲、II期临床CENTAUR研究[8],共纳入137名ALS患者。其中89人被随机分配到苯丁酸钠-牛磺酸二醇组,48人分配至安慰剂组。在改进的意向性治疗分析中,上述药物对ALSFRS-R评分的平均变化为每月-1.24分,安慰剂组为每月-1.66分,两者有统计学意义(见图3)。该研究结果显示苯丁酸钠-牛磺酸二醇比安慰剂延缓了约25%的疾病进展。在CENTAUR 2期临床试验中发现[9],苯丁酸钠-牛磺酸二醇组患者中位生存期为25个月,安慰剂组中位生存期只有18.5个月。同安慰剂组相比,苯丁酸钠-牛磺酸二醇组患者中位生存期延长6.5个月。该研究说明,苯丁酸钠-牛磺酸二醇可有效改善ALS功能恶化并有效延长生存期。 图3 苯丁酸钠-牛磺酸二醇和安慰剂对ALSFRS-R评分影响 研究显示该药安全性和耐受性良好,临床中最常见的副作用是腹泻、腹痛和恶心[8, 9]。苯丁酸钠-牛磺酸二醇于2022年6月获得加拿大卫生部的批准,美国和欧洲的监管机构正在对苯丁酸钠-牛磺酸二醇进行药物审查,有望在更多的地区和国家上市。 4. 氢溴酸右美沙芬-奎尼丁 氢溴酸右美沙芬和硫酸奎尼丁的组合(Nuedexta)被批准用于治疗假性延髓麻痹时强哭强笑症状。假性延髓麻痹也是肌萎缩侧索硬化中相对常见的非运动性症状。右美沙芬是一种非竞争性NMDA-谷氨酸受体拮抗剂,可能具有神经保护特性。硫酸奎尼丁是一种细胞色素P450酶抑制剂,可阻断右美沙芬的首过肝代谢,从而提高右美沙芬的生物利用度。 图4 右美沙芬-奎尼丁对CNS-LS评分的影响 一项来自美国的多中心、随机双盲、对照平行、三臂临床研究发表于Neurology杂志[10],评价氢溴酸右美沙芬-奎尼丁治疗 ALS 假性延髓麻痹中的作用。研究发现联合用药(30毫克右美沙芬+30毫克奎尼丁)组患者较右美沙芬组患者神经病学研究中心-不稳定性量表(CNS-LS)得分降低3.3分,较奎尼丁组患者降低3.7分。该研究说明联合用药比单独服用其中任何一种成分相比,显著降低患者假性延髓麻痹症状。并且该药物耐受性良好,腹泻(13%)和头晕(10%)是常见副作用[10]。 02 超适应征ALS用药 1. 甲钴胺 甲钴胺尚未被批准用于肌萎缩侧索硬化症的治疗;然而今年一项来自日本的多中心、安慰剂对照、随机双盲、III期临床试验发表于JAMA Neurology杂志[11],纳入130名ALS患者,随机分配进甲钴胺组和安慰剂组。接受肌内注射甲钴胺(50毫克/天)或安慰剂,每周两次,持续16周。研究显示16周后甲钴胺组患者ALSFRS-R总分较安慰剂组高1.97分(-2.66 vs -4.63;95%CI 0.44-3.50;P =0.01)。甲钴胺组和安慰剂组不良事件发生率类似。该研究结果说明超大剂量甲钴胺可有效延缓ALS患者的神经功能衰退(见图5)且安全性良好。 图5 甲钴胺和安慰剂对ALSFRS-R评分的影响 2. 盐酸美西律 美西律是一种钠通道阻滞剂,被美国食品和药物管理局(FDA)批准用于治疗心律失常。肌萎缩侧索硬化患者常有肌肉痉挛症状,美西律可减轻下运动神经元钠离子电流的持续增加,减少周围神经的过度兴奋性,从而减轻症状。一项美国的随机、双盲、交叉研究显示,美西律组ALS患者较安慰剂组患者每日肌肉痉挛次数减少1.8次/天[12](见图6)。并且,300毫克美西律被认为是安全且耐受性好。 图6 盐酸美西律和安慰剂对ALS患者肌肉痉挛的影响 03 开发阶段的ALS疗法 1. 小分子制剂 目前诸多小分子制剂在投入研究,这些小分子靶向ALS的各种假说机制,包括兴奋毒性、凋亡、炎症、氧化应激和遗传途径等等。这些小分子正在处于不同的开发阶段,包括I期、II期和III期临床试验。 图7按照作用机制分类针对ALS的在研小分子制剂汇总 例如:CNM-Au8(NCT04414345)[13],一种金纳米晶体,可透过血脑屏障,可通过增加烟酰胺腺嘌呤二核苷酸(NAD)和三磷酸腺苷(ATP),稳定蛋白和减少活性氧来减少可能导致神经元退化的细胞应激反应。另一项针对ALS的多臂、多阶段、适应性试验正在英国进行。MND SMART(NCT04302870)于2022年启动,目前正在测试两种治疗ALS的再利用药物美金刚和曲唑酮[14]。其他针对小分子制剂的研究见上图(图7)。 2. 干细胞 尽管到目前为止,使用各种干细胞的临床试验尚未显示出临床获益,但干细胞作为肌萎缩侧索硬化症的潜在治疗方法正在逐渐成为热点。目前针对干细胞研究主要涉及人神经干细胞和来自骨髓或脂肪组织的间充质干细胞。 到目前为止,大多数干细胞试验规模都很小,而且容易受到研究人群、治疗方案和结果衡量标准之间巨大异质性的限制,这使得结果难以衡量。干细胞治疗的局限性包括经济成本、重复给药的时间密集性、侵入性的治疗途径(鞘内、肌肉内或髓内注射)以及治疗给药所需的技术壁垒等。 3. 基于RNA的干预措施 基于RNA的干预措施以靶向的方式改善致病基因的表达,反义寡核苷酸(ASO)是该种疗法的主要形式。反义寡核苷酸是人工合成的单链分子,长度为8-50个核苷酸,可以与RNA结合并通过多种机制改变蛋白质的表达。其他RNA疗法,如小干扰RNA(SiRNA),可能逐渐在ALS临床试验中被探索。ASO疗法是为遗传性发育和退行性疾病而开发的,它可以被用于治疗遗传型ALS。尽管ASO技术取得了进展,但要取得成功的临床治疗效果,仍然要克服许多障碍,且长期副作用相对未知,需要更多临床试验有助于评估该疗法的长期效果和副作用。 4. 病毒载体 病毒载体可以将DNA传递给异常或功能失调的细胞。最常见的基因治疗载体是腺相关病毒(AAVs),它可以转导神经元来取代缺陷基因,或者沉默突变基因及其蛋白质衍生物。多项针对ALS研究发现,通过AAV递送抑制性microRNAs来减少SOD1蛋白可以延迟ALS动物模型的疾病症状并延长寿命,但目前这些研究正处于临床开发的早期阶段。 目前已经使用不同的递送方式和不同的转基因(例如生长因子如IGF-1或GDNF、mRNA和抗氧化剂)进行了一系列AAVs介导的基因治疗,这些方法增加了运动神经元的存活,改善神经肌肉功能,延迟了症状的出现,并以良好的安全性降低了疾病的进展。病毒载体为ALS的治疗带来了巨大的希望。 5. 单克隆抗体 使用单克隆抗体作为神经退行性疾病的潜在治疗方法逐渐进入研究者视野(例如阿杜卡奴单抗于2021年6月被批准用于治疗阿尔茨海默症)。目前还没有被批准用于ALS的单抗,目前研究阶段单抗汇总见图8。单抗具有多种优点,例如即使在错误折叠状态下也能以高特异性和亲和力靶向蛋白质,半衰期长,并能与其他分子结合。研究的靶点包括SOD1、TDP-43、C9orf72、Nogo-A、肌肉特异性激酶、轴突生长抑制因子、白介素6受体、干扰素、分化簇40配体(CD40L)、结缔组织生长因子、死亡受体6、神经毛细蛋白-1、肌肉生长抑素、高运动组蛋白1和山梨素等等。针对山梨素(AL001)、C1q、SOD1、CD40L和IL-6的单抗目前正在II期临床试验中。该免疫调节疗法对于治疗肌萎缩侧索硬化同样具有令人兴奋的前景。 图8 单克隆抗体治疗ALS临床研究汇总 6. 其他干预措施 非药理途径正在研究中,包括输注来自健康青年的血浆(NCT04454840);脐带血注射(NCT02236065);氨基酸和维生素疗法(NCT03103815);补充剂和食品衍生物干预(烟酰胺NCT04562831);植物衍生物治疗(NCT03690791);人体内自然存在的物质(NCT04518540);益生菌(NCT03324399)和粪便微生物区系移植(NCT03766321)以及饮食疗法等等,这些措施仍然是一门新兴而复杂的科学,距离投入临床使用还有很长的距离要走。


参考文献:
1.Johnson, S.A., et al., Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs, 2022. 82(13): p. 1367-1388.
2.Bensimon, G., L. Lacomblez, and V. Meininger, A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med, 1994. 330(9): p. 585-91.
3.Lacomblez, L., et al., Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet, 1996. 347(9013): p. 1425-31.
4.Andrews, J.A., et al., Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 2020. 21(7-8): p. 509-518.
5.Abe, K., et al., Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener, 2014. 15(7-8): p. 610-7.
6.Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol, 2017. 16(7): p. 505-512.
7.Witzel, S., et al., Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol, 2022. 79(2): p. 121-130.
8.Paganoni, S., et al., Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med, 2020. 383(10): p. 919-930.
9.Paganoni, S., et al., Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve, 2021. 63(1): p. 31-39.
10.Brooks, B.R., et al., Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology, 2004. 63(8): p. 1364-70.
11.Oki, R., et al., Efficacy and Safety of Ultrahigh-Dose Methylcobalamin in Early-Stage Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol, 2022. 79(6): p. 575-583.
12.Oskarsson, B., et al., Mexiletine for muscle cramps in amyotrophic lateral sclerosis: A randomized, double-blind crossover trial. Muscle Nerve, 2018.
13.Vucic, S., et al., Study protocol of RESCUE-ALS: A Phase 2, randomised, double-blind, placebo-controlled study in early symptomatic amyotrophic lateral sclerosis patients to assess bioenergetic catalysis with CNM-Au8 as a mechanism to slow disease progression. BMJ Open, 2021. 11(1): p. e041479.
14.Wong, C., et al., Motor Neuron Disease Systematic Multi-Arm Adaptive Randomised Trial (MND-SMART): a multi-arm, multi-stage, adaptive, platform, phase III randomised, double-blind, placebo-controlled trial of repurposed drugs in motor neuron disease. BMJ Open, 2022. 12(7): p. e064173.

















