
摘要
黏多糖贮积症Ⅰ型是1种由于编码α-L-艾杜糖苷酶的基因变异所致的罕见常染色体隐性遗传病。本病可累及多系统、多器官,临床表现差异较大,病程呈进行性进展,早期诊断、早期治疗可减缓疾病进展,提高患儿生存质量。为规范黏多糖贮积症Ⅰ型的诊疗,中华医学会儿科学分会内分泌遗传代谢学组、中国罕见病联盟和中华儿科杂志编辑委员会基于国内外多项临床研究成果,结合我国临床现状及从业专家的临床经验,共同制定黏多糖贮积症Ⅰ型诊疗专家共识(2022)。
黏多糖贮积症(mucopolysaccharidosis,MPS)Ⅰ型是由于编码α-L-艾杜糖苷酶(a-L-iduronidase,IDUA)的基因变异,导致机体IDUA酶活性缺乏或降低,从而使体内黏多糖(又称糖胺聚糖,glycosaminoglycan,GAG)不能完全降解而贮积在细胞溶酶体中,引起受累组织、器官功能障碍所致的一种溶酶体贮积症,呈常染色体隐性遗传。本病可累及多系统、多器官,临床表现差异较大,病程呈进行性进展,具有较高致残率,严重者未及时治疗可致早期死亡。而早期诊断、早期治疗可减缓疾病进展,提高患儿生活质量。随着酶学、基因检测技术在临床的普及,MPS Ⅰ型的确诊率显著提高,但临床仍存在早期诊断和分型困难以及治疗管理欠规范等问题。为进一步提高医务工作者对本病的认识水平,规范MPS Ⅰ型的诊疗,有效改善患儿预后,中华医学会儿科学分会内分泌遗传代谢学组、中国罕见病联盟和中华儿科杂志编辑委员会于2021年11月始采用德尔菲法,组织相关专家多次讨论,基于国内外多项临床研究成果,结合我国临床现状及从业专家的临床经验,共同制定黏多糖贮积症Ⅰ型诊疗专家共识(以下简称本共识)。
MPS Ⅰ型首次报道于1919年,是最早被发现并报道的MPS亚型。世界范围内MPS Ⅰ型的总体发病率为0.69/10万~1.66/10万,欧美地区发病率相对高于亚洲国家和地区。我国华东、华南地区MPS Ⅰ型在MPS中的占比分别为16%和12%。
依病情严重程度,可将MPS Ⅰ型分为重型(Hurler综合征),中间型(Hurler-Scheie综合征)和轻型(Scheie综合征)。重型也称经典型,多在1岁内发病,中枢神经系统受累明显,有发育落后和倒退;轻型发病偏晚,智力和身高正常;中间型智力可在临界值或者落后,也可正常。一般发病越早,表型越重,病情进展快。无论哪种亚型,均表现为多器官受累的慢性进行性病程。1. 颅面及外观:患儿出生时臀背部、四肢常有大片、多处蒙古斑,2岁左右即可见明显面容粗糙,特征包括皮肤厚,汗毛长;头大,舟型头,头发浓密粗糙,发际低,颈短;前额突出,眉毛浓密,眼睛凸出,眼睑肿胀;鼻梁低平,鼻孔上翻,鼻翼较厚;嘴唇阔而厚,舌大,易伸出口外;牙龈增生,牙齿细小且间距宽;耳垂厚。部分患儿身高早期可能超过同龄儿童平均身高。3岁后,线性生长明显减慢,导致身材矮小。2. 骨骼和关节:患儿均有进行性骨骼发育不良。1岁左右胸腰椎后凸逐渐明显,也可出现其他脊柱异常(脊椎滑脱、齿状突发育不良、脊柱侧凸等)、颅骨异常(J形蝶鞍、颅骨增厚)。大、小关节均可被累及,出现关节活动受限,如肩关节上举受限、髋关节活动受限、膝关节屈曲僵硬、手指弯曲僵硬(爪形手)等。3. 呼吸系统:黏多糖贮积可导致扁桃体和腺样体肥大,气道狭窄,声带增厚,同时舌大。患儿常有慢性复发性鼻炎,睡眠打鼾,慢性阻塞性呼吸暂停,易反复呼吸道感染。由于声音传导性障碍和(或)感音性障碍、慢性中耳炎,患儿常有听力缺陷。4. 消化系统:常见腹部膨隆,肝脾增大,脐疝和腹股沟疝,偶有腹泻和便秘。疝气即使手术修复仍易复发。5. 心血管系统:大部分患儿的心脏累及发生在疾病后期,表现为瓣膜病。黏多糖贮积在二尖瓣、主动脉瓣和三尖瓣,可导致心律失常、左心室肥厚、心力衰竭。少数患儿在疾病早期表现为心肌病和心内膜弹力纤维增生症。随年龄增大,可进展为冠状动脉疾病、肺动脉高压。二尖瓣关闭不全是重型MPS Ⅰ型最常见的瓣膜病。6. 神经系统:重型患儿在1岁左右即表现为智力落后,智力水平为2~4岁,随后缓慢倒退至严重智力障碍。患儿常出现交通性脑积水和腕管综合征,大龄儿童可出现寰枢椎不稳、脊髓受压。7. 眼:患儿早期即有角膜混浊,进展后可出现开角型青光眼、视网膜变性、视神经压迫。视网膜变性可导致周边视野缺失及夜盲症。角膜混浊、视网膜变性、视神经压迫和萎缩、脑积水造成的皮层损伤,可能导致失明。经典型MPS Ⅰ型患儿的自然病史为:出生时大部分有蒙古斑,婴儿早期出现脐疝或腹股沟疝,1岁前反复上呼吸道感染。常于1岁左右出现胸腰椎后凸,进行性骨发育不良及关节病。逐渐表现出严重智力障碍,心肺受累,听力丧失和角膜混浊。中位确诊年龄约为10月龄;如未及时治疗,患儿多于10岁内死亡。
1.IDUA酶活性检测:是MPS Ⅰ型诊断的金标准,IDUA酶活性显著低于正常水平具有确诊意义。检测样本包括外周血白细胞、皮肤成纤维细胞、干血纸片等。检测方法主要为四甲基伞形酮底物荧光定量法或串联质谱法。但应注意IDUA基因的假性缺陷可能导致酶活性检测呈假阳性,需注意排除,应结合尿GAG检测和IDUA基因检测以明确诊断。已知的可致酶活性显著降低的假性IDUA基因变异有c.235G>A、c.246C>G、c.667G>A、c.787A>T、c.965T>A、c.1081G>A、c.1757C>T等。2.IDUA基因检测:主要采用Sanger测序或二代基因测序技术。可通过提取静脉血、干血纸片样本或组织标本的DNA进行基因检测。若发现1个已知致病变异和1个意义未明的变异、2个意义未明的变异,则需结合IDUA酶活性及尿GAG检测等明确诊断。已报道355种IDUA基因变异类型。不同地区及人种IDUA基因变异类型有差异,c.1205G>A、c.208C>T、c.1598C>G多见于大多数欧洲国家以及美洲、澳洲国家。我国MPS Ⅰ型患儿IDUA基因报道较多的变异有c.236 C>T、c.1037T>G。IDUA基因变异类型与表型高度相关。无义变异或移码变异的纯合子或复合杂合子可致IUDA酶活性完全丧失,表现为重型。亚重型患儿通常至少带有1个错义变异。c.1029C>G、c.1960T>G、c.1960T>C、c.590-7G>A变异与亚重型MPS Ⅰ型相关。亚洲人群中较为常见的重型、亚重型相关变异分别为c.1190-1G>A、c.266G>A。3.尿GAG检测:MPS Ⅰ型患儿尿GAG硫酸皮肤素和硫酸类肝素水平可显著增高。尿GAG定量还可作为生物标志物用于造血干细胞移植(hematopoietic stem cell transplantation,HSCT)和酶替代治疗(enzyme replacement therapy,ERT)的治疗效果评估。尿GAG定性检测常用甲苯胺蓝斑点法和醋酸纤维薄膜双向电泳或琼脂糖凝胶电泳法;定量检测常用改良的1,9-二甲基亚甲蓝-Tris比色法进行尿GAG浓度及GAG/肌酐定量分析。4.其他辅助检查:(1)骨骼X线检查包括①肋骨:近脊柱端干骺端增宽呈“括弧状”,远端肋骨明显增宽呈“飘带状”,锁骨近端增宽。②椎体:第2颈椎齿状突发育不良,胸腰椎后侧凸畸形,椎体形状扁平、不规则或前缘鸟喙状异常。③骨盆:髋臼浅、髋外翻、股骨头发育不良。④长骨:骨干变短、不规则,远端增宽,骨髓腔增宽。⑤双手:掌骨近端呈子弹头状,尺桡骨远端呈“V”形。⑥头颅:颅骨增厚,舟状头,蝶鞍J形。(2)头颅CT或磁共振成像典型表现为颅内多发小的囊状改变,其他包括脑室增宽和脑积水等。(3)心电图改变包括心动过速、心肌肥厚、心律失常等。(4)超声心动图可见瓣膜病变(依次为二尖瓣、主动脉瓣、三尖瓣和肺动脉瓣)、心肌肥厚,晚期见充血性心力衰竭等。(5)眼科检查可见不同程度的角膜混浊、眼压增高。(6)听力测试可见不同程度的听力受损。(7)腹部超声检查可见肝脾肿大。
1.诊断流程(图1):基于临床表现、体征或骨骼X线检查疑诊MPS Ⅰ型的患儿,可行尿GAG定性或定量、IDUA酶活性检测、IDUA基因检测,确定致病变异类型。由于二代测序在临床应用的普及,常有疑似患儿先通过二代测序检出IDUA基因变异,如不能确定为双等位基因的致病变异,则需进行IDUA酶活性检测,若IDUA酶活性正常,可排除MPS Ⅰ型。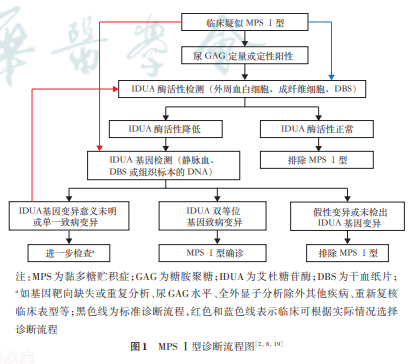
2.鉴别诊断:MPS Ⅰ型主要需与MPS Ⅱ型、MPS Ⅳ A或Ⅳ B型、MPS Ⅵ型、MPS Ⅶ型以及黏脂贮积症Ⅱ型和Ⅲ型等进行鉴别诊断,除MPS Ⅱ型为X连锁隐性遗传外,其余皆为常染色体隐性遗传。特异性酶活性缺乏及致病基因位点的不同是鉴别诊断的关键(表1)。此外,以骨骼改变为著者,尚需与其他遗传性骨病、骨关节病相鉴别。

MPS Ⅰ型治疗方法主要包括HSCT、ERT及对症支持治疗。早诊断、早治疗及多学科管理策略是改善患儿远期预后的关键。确诊后宜尽快行多学科评估,结合基因型及临床表现,明确临床分型,并制定个体化治疗方案。
(一)特异性治疗
1. HSCT:HSCT是重型MPS Ⅰ型患儿的重要治疗手段。通常对年龄≤2.5岁的重型患儿应首选HSCT,以防止认知功能进一步损伤;对年龄>2.5岁的重型患儿或亚重型患儿,经多学科评估移植风险及获益后,亦可考虑HSCT。
MPS Ⅰ型重型患儿经HSCT后远期预后显著改善,国外资料显示HSCT后10年生存率可达83%,可显著降低颈脊髓压迫或严重脑积水等中枢神经系统并发症,其疗效明显优于单纯ERT组及未治疗组。国内研究显示,MPS患儿经HSCT后3年总生存率为84.8%,移植后关节活动度、呼吸道阻塞、肝脾肿大和传导性听力障碍等明显改善。但HSCT对已发生的中枢神经系统损伤、心脏瓣膜病、骨骼病变、角膜混浊、神经性听力障碍等无明显改善作用。随着生存期的延长,部分成年患者可出现抑郁、多动等心理障碍。HSCT亦存在供体来源稀缺、移植物抗宿主病、移植物排斥以及危及生命的严重感染或出血等风险。有关供体选择、预处理方案、移植物抗宿主病的预防等可参考国内外造血干细胞移植相关指南和共识。
2.ERT:重组人α-L-艾杜糖苷酶(注射用拉罗尼酶浓溶液),简称拉罗尼酶,是MPS Ⅰ型的特异性治疗。非重型患儿经ERT长期治疗可缓解全身躯体症状,增加关节活动度、缩小肝脾体积、缓解呼吸道梗阻、稳定心脏瓣膜病和角膜混浊,改善身高增长。但该药物不能透过血脑屏障,不能阻止中枢神经系统进一步损伤。所有MPS Ⅰ型患儿均可从ERT中获益,无明显神经系统受累的非重型患儿获益最大。重型患儿在HSCT等待期及移植后8~12周可考虑ERT联合治疗。
拉罗尼酶的推荐剂量为100 U/kg,生理盐水稀释,缓慢静脉输注,持续3~4 h,每周1次,长期治疗。推荐初始输注速率2 U/(kg·h),如能够耐受,可每15分钟增量1次,直至最大输注速率32 U/(kg·h)。最常见的不良反应为输液相关反应,可通过减缓输注速率、使用抗组胺药和(或)解热剂进行预防或治疗。治疗后应定期随访监测,评估治疗效果(表2)。

(二)对症治疗
MPS Ⅰ型常累及多系统、多器官,呈进行性加重。一旦诊断,应组织多学科专家参与评估和管理。进行性的脑积水、颈脊髓压迫、脊柱弯曲、髋关节发育不良、心脏瓣膜病、角膜混浊等并发症,需要进行脑积水分流术、脊髓压迫松解术、脊柱矫形、髋关节矫形术、心脏瓣膜置换术、角膜移植等对症干预。脊柱后凸是MPS Ⅰ型患儿常见表现,早期可给予支具、止痛等治疗,必要时于儿童期(5~13岁)手术矫正。少数患儿在HSCT及ERT后角膜混浊仍进行性加重,在排除视网膜病及视神经病变后可考虑角膜移植。反复发作的中耳炎可行鼓膜置管术。斜疝、腺样体肥大、呼吸道梗阻等给予对症治疗。
MPS Ⅰ型呈常染色体隐性遗传,对先证者及其直系亲属应进行遗传咨询,评估患病风险。高危家庭再生育时可进行产前诊断,常于妊娠10~13周取胎盘绒毛或16~22周取羊水细胞进行IDUA酶活性测定及IDUA基因变异分析,也可进行胚胎植入前遗传学检测。少数国家或地区已将MPS Ⅰ型纳入新生儿筛查计划,常采用串联质谱法或荧光底物法测定新生儿干血斑IDUA酶活性。MPSⅠ型是一种常染色体隐性遗传溶酶体贮积症,临床可累及多系统、多器官,临床表现差异较大,病程呈进行性进展。诊断需基于临床表现、体征、骨骼X线检查,行尿GAG定性或定量分析、酶活性检测、基因分析。早期诊断、早期治疗可减缓疾病进展,提高患儿生存质量。

















