据估计,欧盟现有5000~8000种罕见病。虽然罕见病的发病率较低,并且大多数不超过1/10000,但其影响欧盟2700~3600万人,占总人口的6%~8%。因此,对于公共卫生政策制定者来说,孤儿药的发展则是其致力于满足患者需求的一个重要考虑因素。对制药企业来说,在正常的市场条件下,开发和销售孤儿药通常是不能盈利的。因此,欧盟为孤儿药的研发者提供一定的激励,在孤儿药认定和上市许可方面构建了较为完善的管理机制。自2000年欧盟孤儿药法规实施至2015年年底,欧盟认定了1596个孤儿药,上市许可121个孤儿药。本文在对欧盟孤儿药法规政策分析的基础上,系统分析2000-2015年欧盟孤儿药认定和上市许可的情况,为我国孤儿药政策法规制定和孤儿药研发提供参考。
1、欧盟孤儿药政策分析
孤儿药法规的主要目的是确保罕见病患者与任何其他疾病患者享有同等的治疗质量,内容主要包括制定孤儿药认定程序、制定已认定的孤儿药研发与上市的激励政策、成立孤儿药委员会。还包括鼓励各成员国在国家层面上有类似的和/或补充政策。欧洲药品监督管理局则通过COMP审核申办者的孤儿药认定申请,并通过免费的申请提交前会议协助申办者准备孤儿药认定申请,及对孤儿药认定后的研发提供建议。
1.1孤儿药认定标准及程序
欧盟孤儿药的认定基于孤儿药法规制定的标准。获得孤儿药认定资格,需满足以下条件:1)必须用于治疗、 预防或诊断危及生命或慢性衰弱的疾病。2)在欧盟的发病率不超过5/100000或药品上市销售收入不可能匹敌其研发所需的投入。3)没有令人满意的诊断、预防或治疗相关疾病的方法被批准,或如果这样的方法存在,但相关药物的显著效益受到相关疾病的影响。
孤儿药认定是免费的,可在上市许可前的任何研发阶段申请,需提供治疗作用的科学合理性依据。COMP通过其已建立的专家网来审查孤儿药认定申请。孤儿药认定程序包括:1)申办者告知EMA其提交申请的意愿。2)申请提交前的会议电话会议。3)提交孤儿药认定申请。EMA确认申请(1d)。4)评估COMP会议/可能有的口头审理/COMP给出意见(自确认起60或90d)。5)意见发送给欧盟委员会。6)EC批准孤儿药资格(30d内)。7)认定结果发布在EC网站的孤儿药公共注册平台,同时,孤儿药认定意见的公告发布在EMA网站。
1.2孤儿药认定后的行动
获得孤儿药认定的申办者可得益于许多孤儿药激励。包括方案指导,一旦孤儿药上市就获得一定期限的市场排他权及根据申办者的情况而进行的费用减免等,具体分析见“1.3” 但获得孤儿药认定并不表明其已经满足了上市许可批准所必需的有效性、安全性和质量标准。与任何药物一样,只有提交上市许可申请之后才能评估这些标准。
获得孤儿药认定后,申办者需每年向EMA提交一份关于该孤儿药研发状况的年度总结报告。获得认定的孤儿药的上市许可申请由人用药品委员会进行评估。同时,申办者也需要提交一个维持其孤儿药认定资格的申请,以获得10年市场排他权激励。孤儿药认定可以从一申办者转让给另一个申办者,并且是免费的。在申办者的要求,或孤儿药上市许可前不再符合标准,或市场排他权期满的情况下,认定的孤儿药可被撤销资格。
1.3孤儿药研发激励政策
在欧盟,法律为激励申办者/制药企业研发孤儿药提供政策保障。通过孤儿药认定程序认定的药物有资格获得相应激励,1)方案指导:EMA为孤儿药研发方案的优化提供科学建议及申请文件的准备进行指导,以满足EMA监管要求。申办者的方案指导要求没有次数限制。这有助于申办者最大化其孤儿药上市许可申请获得批准的机会。2000-2015年EMA共完成951个(次)方案指导,其中264个(次)涉及中小型企业。2)集中许可程序:在欧盟,所有认定后的孤儿药的上市许可申请都进入集中许可程序。允许制药企业只向EMA提交单一申请,EC的意见和决策在所有欧盟成员国有效。目前,绝大多数创新药通过此方式快速在欧盟上市。3)条件上市许可:基于不全面的数据来批准孤儿药,以解决未满足的医疗需求。这些数据必须表明该孤儿药的收益远远大于它的风险,及申请人以后有能力提供全面的临床数据。4)市场排他权:孤儿药上市许可获得批准之后,可获得10年的欧盟市场排他权。在此期间,直接竞争类似产品不能上市。如果在孤儿药认定审查期间,遵守儿科试验计划协议,市场排他权期限可再延长2年。目前有2个儿童用孤儿药获得12年市场排他权。5)对中小型企业的额外激励:中小型企业研发孤儿药可获得进一步的激励,如来自EMA中小型企业办公室的行政和程序上的帮助,还可以获得一定的费用减免费用减免:对获得孤儿药认定的企业的监管活动进行费用减免,包括上市许可前的方案指导、上市许可申请和检查费用,上市许可后的申请变更费用及年费,中小型企业的额外减免费用等。根据欧盟预算,费用减免情况每年修订。2000-2015年费用减免共计约7840万欧元。7)欧盟的研究资助:研发孤儿药的申办者有资格获得来自欧盟和成员国的研发项目资助和活动支持。 EMA不给孤儿药的申办者提供研究资助,资助可从EC和其他渠道获得。计划在美国和日本进行孤儿药研发的申办者也可以获得资助。相关激励政策的法律来源见表1。
2、2000—2015年欧盟孤儿药认定情况分析
近年来,向EMA递交的孤儿药认定申请的数量显著增加。制药企业对孤儿药认定过程的持续兴趣预示将会有更多的孤儿药上市。
2.1 数量
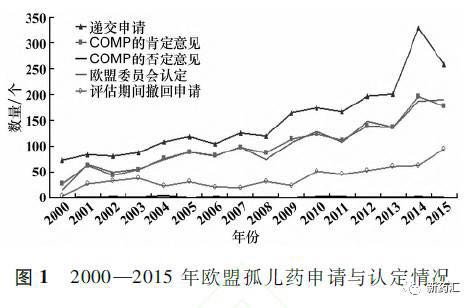
据统计2000-2015年,EMA共计收到2385个孤儿药认定申请;COMP对1607个申请给予肯定意见,21个申请给予否定意见;EC认定1596个孤儿药资格;评估期间申办者撤回607个申请。大多数孤儿药认定基于普遍标准,1个孤儿药认定基于“投资回报不足”的标准。目前,1237个孤儿药资格是有效的。具体情况见图1。从发病率分布情况来看:<1/10000(640个),约占40%;≥1/10000,≤3/10000(786个),约占49%;>3/10000(170个),约占11%。
2.2治疗领域分布
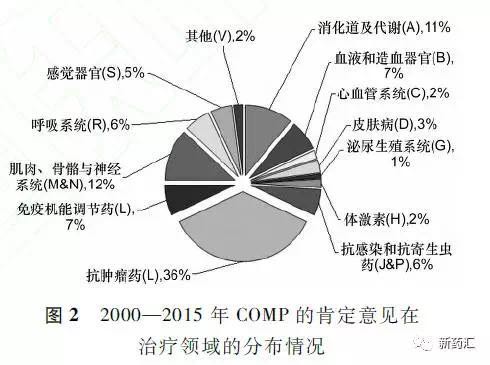
据统计2000-2015年COMP的1607个孤儿药肯定意见在治疗领域的分布情况依次为:抗肿瘤药579个,约占36%;肌肉、骨骼与神经系统用药193个,约占12%;消化道及代谢用药177个,约占11%。具体情况见图2
2.3治疗对象分类
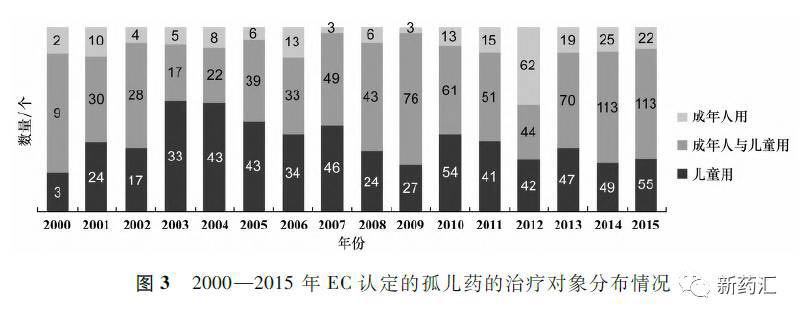
据统计,2000-2015年EC认定的1596个孤儿药中儿童用药为216个,约占14%;成年人用药582个,约占36%;成年人与儿童共用药798个,约,50%。 具体情况见图3
3、2000-2015年欧盟孤儿药上市许可情况分析
3.1数量
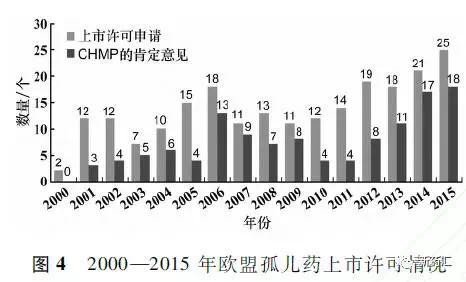
据统计,2000-2015年EMA共计收到认定后的孤儿药的上市许可申请220个,获得CHMP推荐上市的孤儿药121个,其中82%是新活性物质,25个上市许可孤儿药来自中小型企业。具体情况见图4。从发病率分布情况来看:<1/10000(58个),约占48%;≥1/10000,<2/10000(33个),约占27%;≥2/10000,小于3/10000(16个)。约占13%;≥3/10000(14个),约占12%。
3.2治疗领域分布
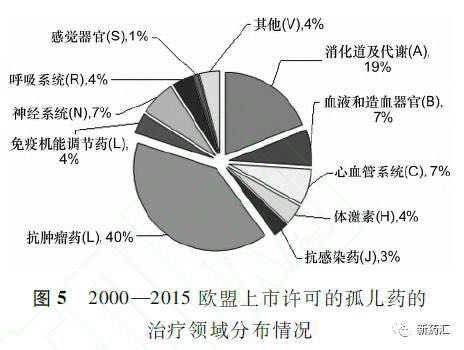
据统计,2000-2015年欧盟上市许可的121个孤儿药的治疗领域分布情况依次为:抗肿瘤药49个,约占40%;消化道及代谢用药23个,约占19%;血液和造血器官用药8个、心血管系统用药8个、神经系统用药8个,各占7%。居体情况见图5。
3.3药物的类型分布
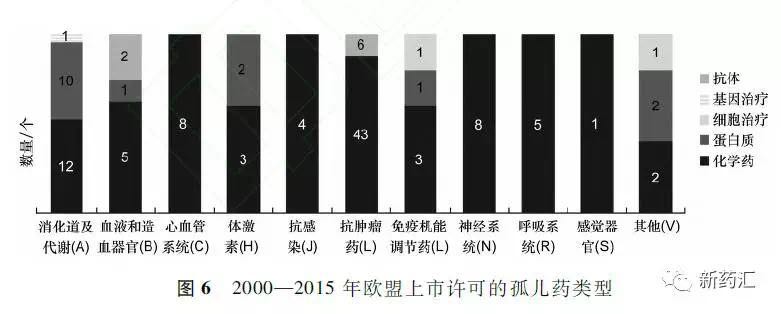
据统计,2000-2015年欧盟上市许可的121个孤儿药的药物类型分布情况,化学药94个,约占78%;蛋白质类药16个,约占13%;抗体类药物8个,约占7%。具体情况见图6。
4、结论
到目前为止,欧盟的孤儿立法是成功的2000-2015年,孤儿药认定成功率67%,孤儿药上市许可成功率55%。 虽然孤儿药认定和上市许可的前景是光明的,但令人担心的是,因为高昂的治疗成本,在欧盟并非所有罕见病患者都能获得这些药物。EMA一直在考虑如何提高其孤儿药科学决策的透明度,以便欧盟成员国的药品定价机构在做出孤儿药定价决策时可以参考。因为罕见病是一个全球性的问题,为罕见疾病患者提供更多更好的治疗,则需要全球药品监管机构之间的紧密合作。孤儿药的未来机遇与挑战并存。
来源:新药述评与论坛

















