今日,美国FDA宣布,加速批准Sarepta Therapeutics公司的Vyondys 53(golodirsen)上市,治疗适于使用外显子53跳跃(exon 53 skipping)治疗的杜兴氏肌营养不良症(DMD)患者。这些患者大约占DMD患者总数的8%。Sarepta公司曾在今年8月受到FDA关于golodirsen上市申请的CRL,不过这款创新疗法今天终于冲过了终点线!
DMD是由于在X染色体上编码抗肌萎缩蛋白的基因上出现突变而导致的罕见遗传病。抗肌萎缩蛋白的缺失或缺陷,导致肌肉在收缩过程中出现慢性损伤,炎症发作,影响肌肉的再生。最终,肌肉被瘢痕组织或者脂肪代替。患者的肌无力症状在2-3岁时就很明显,随着肌肉组织和功能的不断丢失,在12岁时通常只能靠轮椅行动,20岁时需要辅助呼吸,在30-40岁时因为呼吸或心力衰竭而早夭。
目前,大部分现有疗法只能缓解疾病症状,却不能针对疾病根源,DMD患者的希望在于创新疗法的开发。

通过外显子跳跃可以治疗大部分DMD患者(图片来源:Sarepta官网)
Vyondys 53是一种Sarepta开发的一种磷酰二胺吗啉代寡聚核苷酸。它靶向抗肌萎缩蛋白mRNA前体的剪接过程,引入外显子53跳跃,旨在产出截短但仍具有功能的抗肌萎缩蛋白。这款疗法获得FDA授予的快速通道资格,孤儿药资格和优先审评资格。它的获批让Sarepta公司获得一张罕见儿童疾病优先审评券(rare pediatric disease priority review voucher)。
Vyondys 53的疗效在一项包括两部分的临床研究中得到评估。第一部分包括12名DMD患者,8名患者接受Vyondys 53的治疗,4名接受安慰剂治疗。第二部分为开放标签试验,包括第一部分的12名患者和13名新的DMD患者。在这项研究中,接受>48周治疗后,患者平均肌萎缩蛋白水平从正常水平的0.1%(基线)提高的正常水平的1.02%。
Vyondys 53的加速批准是基于接受治疗的患者骨骼肌中的肌萎缩蛋白水平升高的替代终点。FDA认为,申请中肌萎缩蛋白水平升高有可能和力预测DMD患者的临床效益。目前,这一疗法的临床效益还未得到确认。作为加速批准的一部分,FDA要求Sarepta公司进行临床试验,验证这一疗法的临床效益。目前正在进行的临床研究将评估Vyondys 53能否条DMD患者的运动功能。如果临床试验不能够证实临床效益,FDA可能收回对这一药物的批准。
“FDA认识到严重神经疾病患者的急迫需求。我们长久以来致力于与研究人员,医药公司和患者合作,促进对罕见疾病疗法的开发。今天的加速批准,让携带特定基因突变的DMD患者首次获得靶向这些基因突变的治疗选择,”FDA药品评估和研究中心神经科学办公室代理主任Billy Dunn医学博士说:“使用加速批准,能够基于初步数据,将Vyondys 53早日交给患者。我们期待从正在进行的验证性临床试验中了解这一疗法的更多临床益处。”
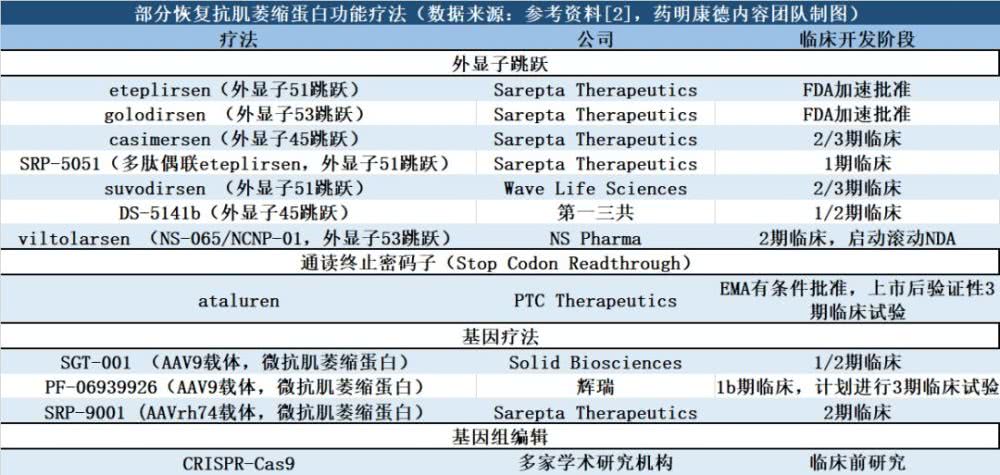
附录:部分恢复肌萎缩蛋白功能的在研和获批疗法
参考资料:
[1] FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation. Retrieved December 12, 2019, from https://www.prnewswire.com/news-releases/fda-grants-accelerated-approval-to-first-targeted-treatment-for-rare-duchenne-muscular-dystrophy-mutation-300974396.html
[2] Verhaart and Artsma-Rus. (2019). Therapeutic developments for Duchenne muscular dystrophy. Nature Reviews Neurology, https://doi.org/10.1038/s41582-019-0203-3

















