多发性硬化症(MS)的疾病修正疗法(DMT)使患者的生存期更长,但这也给患者带来了更高的罹患神经退行性病的风险。MS患者出现的肌肉萎缩,或预示着患者合并发生了其他类型的疾病。本文报导的病例是一名52岁的女性患者,自1999年以来确诊为复发缓解型硬化症(RRMS),最近患者的临床症状迅速恶化,表现为进行性的延髓功能障碍、全身性肌肉萎缩、上运动神经元受累症状。随后的神经生理学检查结果证实了患者合并有肌萎缩性侧索硬化症(ALS)。通过该病例,我们探讨了这两种症状相似且似乎有内在关联的疾病,强调了在MS患者疾病进展后评估是否合并有ALS的必要性。

病例描述
患者女性,52岁,自1999年起定期接受RRMS随访。多年来经历了多次MS复发,使用过β-干扰素、特利氟米特、芬戈利莫德等来控制病情。2018年,在用芬戈里莫德治疗(DMT疗法之一)后,患者的MS没有再复发。自发病以来的多次MRI扫描均证实了患者病情反复和累积性的白质病变(图1)。眼科OCT检查提示双侧视神经萎缩变性。同年,由于认知力下降,患者在接受神经心理学评估时发现其短时记忆障碍。同时期的神经系统检查发现其双侧视神经盘苍白,右眼持续眼球震颤,左半身4级偏瘫,左侧肢体轻度共济失调(EDSS4.0),虽然她的自主行动能力尚无大碍。
图1 患者2014年至2020年的水平位T2-FLAIR扫描显示白质病变的累积。
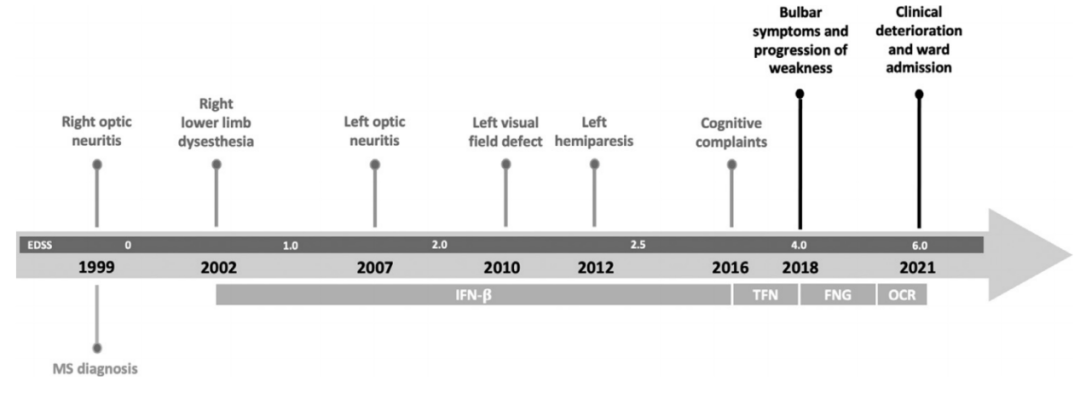
2018年,患者出现了进行性加重的延髓受累表现,如构音障碍、吞咽困难等。但反复的神经影像学评估未发现明确的MS病变(图2),因此诊断为继发性进展性MS。2020年5月,患者开始使用奥克雷珠单抗,到2021年6月患者的上、下肢无力症状又有加重(EDSS 6.0),遂来我院就医(病情发展和治疗示意见图3)。
图2 脑和脊髓MRI。矢状位的T2-FLAIR扫描显示胼胝体萎缩(图A,绿色区域),未发现新的脑干病变(A,箭头);病变主要影响脑室周围白质(Dawson指)(图B,虚线箭头);矢状面T2加权扫描排除了颈椎束中有新的脱髓鞘病变发生(图C)。 图3 疾病发展和诊断治疗的时间线:EDSS:扩展的伤残状态量表。IFN-β:β干扰素。TFN: 特利氟米特。FNG: 芬戈利莫德。OCR: 奥克雷珠单抗。 入院时,她面容憔悴,肢体无力,只能通过平板电脑进行交流,且有蹒跚步态、咳嗽无力、反式呼吸、严重流涎、情绪不稳定的表现。体格检查发现:患者舌肌萎缩、颈部肌肉和上下肢肌肉均无力且萎缩明显,下肢还有肌痉挛表现。肌力评估:右上肢瘫痪,MRC2级,左上肢MRC3级,下肢为4级。患者深感觉、浅感觉均正常,四肢腱反射尚灵敏(3+),足底反射为屈曲。 检测患者的视觉诱发电位发现,其视神经可能存在双侧基底节前神经传导障碍,而其听觉和体感诱发电位检测未提示异常。周围神经传导无异常,针极肌电图显示运动单元明显减少,舌肌有神经再形成的迹象,慢性神经源性运动单位的电位较高。双上肢和下肢近端和远端肌肉出现急性去神经化的表现,如肌肉颤动和肌电尖波。 在对患者做了NGS测序(包括运动神经元疾病相关的的119个基因,其中82个为核基因,37个为线粒体基因),并结合DNA的半定量分析、聚合酶链反应(PCR)、实时qPCR和多重结扎依赖探针扩增(MLPA)后发现:C9orf72基因扩增阴性,但位于15q21.1上SPG11基因的两个意义不明(VUS)杂合变异通过了筛选:SPG11 (NM_025137.3) c.4669G>A(p.(Glu1557Lys))和SPG11(NM_025137.3)c.6877C>T(p.(Arg2293Trp))。前者为内含子变异,后者为错义变异,结果预测为功能丧失可能。且这两种变异在ALS 5型、痉挛性截瘫11型(SPG11)和CMT2X(Charcot-Marie-Tooth disease type 2X)的患者中都有被报道过。 综上,患者被诊断为ALS,并开始用利鲁唑治疗,后又开始用无创通气辅助呼吸,经皮内窥镜胃造口术进食等。此后,患者在家里接受物理和药物治疗,然而病情仍在不断恶化,最终于2022年5月去世。 讨论 DMT疗法显著延长了MS患者的生存期,但也为之带来了更高的罹患衰老相关的神经退行性病的风险。MS患者的肌肉萎缩,或是因为皮质脊髓束功能障碍、不活动和肌肉失用共同所致。然而,也有一些研究证实MS和ALS之间确有联系。已知C9orf72基因的重复扩增可导致脑白质脱髓鞘病变,而少数ALS患者的C9orf72基因也有六核苷酸重复扩增。一个包含了650名ALS患者入组的队列研究发现,有5名ALS患者同时患有MS(其中4位有C9orf72基因核苷酸重复扩增)。与我们的这例患者类似,文献报道的病例大都在确诊ALS之前被诊断为MS,且其中大多数是RRMS—这显然是因为MS起病年龄较小。多数患者始于肢体肌肉萎缩和肌无力,只有少数病例有横纹肌病变症状。 本病例或证明了一个不幸的巧合,或又是MS和ALS之间联系的一个例证。MS被公认为是一种神经炎性病,而ALS则被认为是一种神经退行性病。但越来越多的证据表明,ALS的神经退行性标志物如线粒体功能障碍、氧化应激和细胞骨架蛋白代谢障碍也与MS患者的病情进展有关。同时,本病例的遗传学检测结果提示SPG11的致病可能性。已有研究表明,SPG11基因敲除小鼠出现了早发性运动功能障碍和认知异常,与ALS模型动物的表型相似。从临床表现上来说,两者都会出现四肢瘫;从神经病理学上来讲,蛋白泛素化异常、p62聚集和运动神经元变性也是ALS的标志;而且SPG11基因变异也被认为是青少年ALS的遗传因素之一。SPG11基因编码spatacsin,命名源自“痉挛伴胼胝体变薄或萎缩症蛋白”,虽然生信报告提示该突变是VUS,但这一发现或会激发对SPG11表型及它与MS关系的讨论,虽然目前二者的关联并不那么直接。